
Hastalıkların genetik temeline ait inanılmayacak kadar çok yeni bilgiler ortaya çıkmaktadır. Bu bilgi patlaması polimeraz zincir reaksiyonu (PCR) ve Otomatik DNA dizi analizi ile ateşlenmiş ve insan genom projesindeki hızlı ilerlemeler ile körüklenmiştir. Çok büyük umutlar vermesine rağmen genetiğin günlük tıbbi pratiğe entegrasyonu zorlu bir uğraş olarak kalacaktır. Şimdiye dek, genetiğin en önemli etkisi hastalığın etiyoloji ve patogeneziniîı anlaşılmasını sağlamak olmuştur. Yakın zamanda hastalığın tanısında. Önlenmesinde ve tedavisinde genetiğin çok büyük rol oynayacağını bekleyebiliriz.
Genetik bozukluklar, genellikle bilinenden daha yaygındır, Örneğin, gebeliklerin %3’üntin genetik hastalıklı ya da doğum hasarlı bir bebekle sonuçlandığı tahmin edilir. Bütün pediatrik ve erişkin hasiuııe yatanlarının hemen hemen %10’ u genetik hastalıkla ilişkilidir, Eğer diyabet ve kardiyiovasküler hastalıklar gibi kompleks multifaktöriyel genetik hastalıkları da dahil ederseniz bu rakam yükselecektir. Genetik hastalıkların prevalansı, ağır seyretmesi ve kronik özelliği ile birleştirildiğinde topluma ekonomik, sosyal ve duygusal bir yük getirir.
Kromozomal ve metabolik hastalıklarla ilgili teşhis teknikler mevcut olduğu için, genetik tarihi olarak uzun süre bunlar üzerine odaklanmıştır. Örneğin, trisomi 21 (Down sendromu) ya da monozomi X (Turner sendromu) gibi durumlar sitogenetikle teşhis edilebilir. Benzer şekilde, çoğu metabolik hastalıklar (fenilketonuri, ailesel hiperkolesteromi v.b.) biyokimyasal analizler kullanılarak teşhis, edilmektedir. DNA teşhislerindeki son gelişmeler, genetiğin alanını tıbbın hemen hemen bütün dallarını kapsayacak şekilde genişletmiştir. Örneğin, kardiyolojide kalıtsal karydiyomiyopatilerin ve aritmilere neden olan iyon kanalı kusurlarının mole- küler temeli tanımlanmıştır. Nörolojide, genetik şaşırtıcı sayıdaki nörodejeneratif hastalıkların patofizyolojisini açıklamıştır. Hematoloji, hemoglobinopatilerin başlangıçtaki tarif edilişlerinden bugünkü eritrosit hücresi membran defektlerinin, pıhtılaşma bozuklukları ve trombotik bozuklukların moleküler temelinin anlaşılmasına kadar çarpıcı bir şekilde gelişmiştir. Şimdi neoplazi ve metastatik potansiyel kazanımın genetik olarak tanımlanabildiği çok açıktır.
Genetik çalışmalardan kaynaklanan yeni kavramlar, bazen daha önce aydınlanmamış olan bazı konuların açığa çıkmasını sağlar. Örneğin, çok farklı genetik defektler, periferik nöropatilere sebep olduğu halde, miyelin kılıfın normal durumunun bozulması genellikle son yoldur. Obezitenin çok sayıda genetik sebepleri, iştah için anahtar mekanizma olan proopiomelanocortin polipeptidi ve MC4R reseptörü ürününü içeren fizyolojik bir yolda birleşiyor gibi görünmektedir. Benzer durum Alzheimer hastalığının genetik olarak farklı formları için de ortaya çıkar; bunların bir kaçı nörofibriller dolaşıklık formasyonuna yol açar. Defektif genlerin gittikçe artan oranda tanımlanması anahtar fizyolojik işleme katüan hücresel yolu belirler. Örnekler, kistik fibrozis iletim regülatör (CFTR) genini, Duchenne müsküler distrofi genini (DMD) ve akondroplastik cücelikten sorumlu fibroblast büyüme faktörü (FGFR3) geninin tanınmasını kapsar. Benzer şekilde, transgenik ve ’’knockout” gen modelleri genlerin fizyolojik olarak fonksiyonlarının açığa çıkmasına yardım eder. Genetik yaklaşımlar enfeksiyöz patojenlerin saptanmasında paha biçilmez öneme sahip olduğunu göstermiş ye mîkobakteri, virüs ve parazitler gibi kültürü zor olan ajanların klinik olarak belirlenmesinde kullanılmıştır. Çoğu durumlarda, moleküler genetiğin diyagnostik testlerin doğruluğunu ve uygunluğunu güçlendirmesi, fizyopatalojiyi anlamamızı sağlamış ve gen terapide dahil, terapi için yeni ufuklar açmaya başlamıştır.
Gittikçe daha çok açığa çıkmaktadır ki, genetik altyapı hemen hemen bütün tıbbı olaylarda rol oynamaktadır. Bu, hastalığa karşı duyarlılık, genetik alt yapı ile çevre arasındaki ilişkiler ve hastalıklara, farmakolojik ajnn ya da ilaçların metabolizmasına karşı konak yanıtlan dikkate alındığında, özellikle doğrudur. Genetiğe geleneksel olarak nispeten nadir görülen tek gen hastalıklarının penceresinden bakılmasına karşın, hipertansiyon; astım, diyabet, kalp damar hastalıklanna duyarlılık ve mental hastalıklar, genellikle hastanın aile öyküsünden anlaşıldığı gibi, genetik alt yapıdan da etkilenir. Bu kompleks genetik özellikler, hastalık riskini etkileyen çevre faktörleri kadar, çok çeşitli genlerin katkısıyla oluşur.
Şaşırtıcı oranda ortaya çıkan genetik bilgi, hekimler ve diğer sağlık çalışanları için büyük bir meydan okuma olarak ortaya çıkar. Keşif için kullanılan terminoloji ve teknikler devamlı olarak gelişmektedir. Çoğu genetik bilgi, halihazırda bilgisayarlara yüklenmiş ya da temel bilimsel dergilerde basılmıştır. Biyoenformatikteki gelişmeler, görünüşte göz korkutucu olan bilgi hücumunu basitleştirecektir. örneğin şimdi, organ sistemi, hastalık durumu ya da genlere uygun bağlantı yolu ile ulaşılabilen bir ”web" adresi (http://www.genlirik.wustl.edu) yoluyla genetik test merkezlerine ulaşmak olasıdır. Monogenik bozukluklar "Online Mendelian Inheritance in Man (OMlM; Home - OMIM - NCBI)'de sürekli olarak geliştirilen geniş özetler halinde toparlanmıştır. Bı) ve diğer veri tabanları (http://www.genebank.com) insan genom projesindeki (HGP) gelişmelerle bağlantılı olarak genişletilecektir.
KROMOZOMLAR VE DNA REPLİK ASYONU
DNA’NIN KROMOZOM ŞEKLİNDE ORGANİZASYONU

İnsan Genomunun Büyüklüğü: İnsan genomu 22 otozom (1 den 22’ye kadar numaralanmış) ve X, Y seks kromozomlarım içeren 23 farklı kromozoma bölünmüştür. Erişkin hücreler diploittir; yani iki homologa sahip 22 otozom ve bir çift seks kromozomunu içerirler. Dişiler 2X kromozomu (XX), buna karşılık erkekler bir X ve bir Y kromozomuna (XY) sahiptir. Mayozun sonucu olarak, germ hücreleri (sperm ya da oositler) haploittir ve 22 otozomluk bir takım ve seks kromozomlarından birisini içerir. Fertilizasyon esnasında, anneden ve babadan gelen homolog kromozomların çift halinde yeniden düzenlenmes ile diploit genom oluşur. Her hücre bölünmesiyle (mitoz), kromozomlar replike olur, eşleşir, dağılır ve iki yavru hücreye ayrılır.
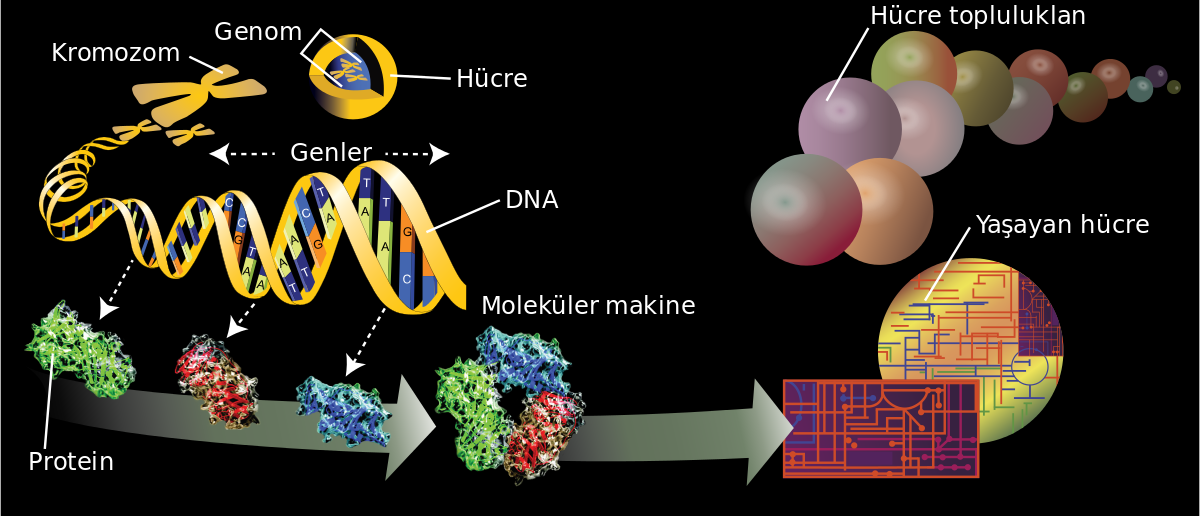
Genomun 23 kromozom arasında bölünmüş 100.000 kadar gen içerdiği tahmin edilmektedir. Bir gen, transkripsiyonla kontrol edilen (aşağıya bakınız) ve hücre içindeki bir aktivitede kullanılacak olan RNA ya da protein gibi bir ürünü kodlayan fonksiyonel bir ünitedir. Târihi olarak genler, bir generasyondan diğerine aktarmakla sorunılu oldukları özelliklere göre kimliklendirilmişlerdir, insan DNA’ sının bir haploit genom için 3 milyar baz çifti (bç) kadar DNA’ dan oluştuğu tahmin edilmektedir. DNA’nın uzunluğu normal olarak 1000 baz çiftlik (kilobaz, kb) ya da 1.000.000 baz çiftlik (megabaz, Mb) birimlerle ölçülmektedir. DNA’nın hepsi genleri kodlamaz. Aslında, genler DNA’nın sadece yaklaşık %10- 15’i kadarda. DNA’nın geri kalan kısmının çoğu fonksiyonları çok az anlaşılmış olan tekrarlayan dizilerden oluşmuştur. Bu tekrarlayan DNA bölgeleri, gen kodlamayan tekrarlamayan dizilerle birlikte DNA’nın kromatin ve kromozom şeklinde paketlenmesinde yapısal rol oynarlar (DNA histon proteinlerine bağlanır) (Şekil 65.1). Eğer DNA’ mn %10’u eksprese oluyor ve 100.000 gen var ise bir gen ortalama 3 kb uzunlukta olacaktır. Bir çok gen bu uzunlukta olduğu halde yayılım oldukça geniştir. Örneğin, bazı genler sadece birkaç yüz baz çifti uzunlukta iken DMD geni gibi genler inanılmayacak kadar büyüktür (2 milyon baz çifti).
DNA’nın yapısı Her gen lineer polimer bir DNA parçasından oluşmuştur. DNA dört farklı bazdan oluşan çift iplikli bir helikstir: Adenin (A), Timin (T), Guanin (G) ve Sitozin ( C). Çift iplikli he-liksi birbirine bağlayan hidrojen bağları sayesinde adenin timin ile guanin si- tozin ile eşleşir. DNA genetik bilginin aktanlmasmı ideal yapan dikkate değer özelliklere sahiptir. RNA veya proteinlerle karşılaştırıldığında oransal olarak da kararlıdır (stabildir). DNA’nın çift iplikli yapısal özelliği ve komplementer baz eşleşme özelliğine bağlı kalışı, hücre bölünmesi esnasında replikasyonun doğru yapılmasını sağlar. Yukarıda belirtildiği gibi komplementerlik özelliği, genetik bilginin DNA JE RNA yEprote- in şeklinde aktanlmasmı da sağlar. Haberci (mesenjer) RNA (mRNA) DNA çift heliksinin anlamlı denilen ipliğinin aynısı olarak kodlanır ve proteinlere translasyonu ribozomlarda yapılır.
Dört farklı bazın oluşu çok fazla genetik çeşitlilik sağlar. Genlerin protein kodlayan bölgelerinde DNA bazları kodonlar şeklinde düzenlenmişlerdir. Baz tripletleri belli bir amino asiti belirler. Dört bazı, 64 değişik triplet kodon (43) halinde düzenlemek mümkündür Her kodon 20 farklı amino asitten birisini ya da translasyonu durdurucu stop kodonu gibi düzenleyici bir sinyali belirler, Amino asitlerden daha çok kodon olması nedeni ile genetik kod dejeneredir yani çoğu amino asitler birden fazla farklı kodonla belirlenirler. Kodonları değişik kombinasyonlarda ve değişik uzunluklarda düzenleyerek çök değişik primer protein yapısı meydana getirmek mümkündür.

Mitozdan hemen önce, hücreler dinlenme ya da Go durumun1 dan çıkar, hücre döngüsüne girerler. Kritik G1 kontrol noktasını geçtikten sonra, hücreler DNA sentezi (S fazı) yaparlar) bu esnada kardeş kromatidler ikişer çift oluşturarak (2n—>4n) hef kromozomun DNA’sı replike olur. Gelecek generasyonlann hücrelerine yanlış aktarmayı önlemek için DNA sentezinin kesinlikle doğru olması gerekir. Diğerleri arasında Xeroderma pigmentosum. Bloom sendromu, ataksi telangiektazi ve kalıtsal nonpolipoz kolon kanseri (HNPCC) DNA yanlış eşleşme genetik anomalilerine dahildir. Bu hastalıkların çoğu, ilave mutasyonlann hızla oluşumunu sağlayarak neoplaziye zemin hazırlar . DNA sentezinin tamamlanmasından sonra, hücreler G2 fazına girerler ve mitoza girmeden önce ikinci kontrol noktasına doğru ilerler. Bu safhada, kromozomlar kondanse olur ve metafazda ekvator bölgesi boyunca dizilirler, Sentromerde birbirine bağlı olan iki identik kardeş kro- matid ayrılırak bölünür ve hücrein iki zıt kutbuna doğru göç eder. Ayrılmış iki takım kromatid etrafında nükleer zar oluştuktan sonra, hücreler bölünür ve diploit (2n) duruma sahip iki yavru hücre meydana gelir.
MAYOZ ESNASINDA GENLERİN AYRILMASI VE DÜZENLENMESİ
Mayoz sadece gonadların germ hücrelerinde meydana gelir. Mitozla bazı özellikleri paylaşır fakat kromozom sayısını haploit sayıya indiren iki belirgin hücre bölünmesi basamağı vardır. Ayrıca genetik çeşitliliği sağlayan aktif bir rekombinasyon vardır. Birinci hücre bölünmesi esnasında, her bir kromozom çifti için iki kardeş kromatid oluşur (2n —» 4n) ve homolog maternal kromozomlar arasındar DNA değişimi vardır, Bu oluşum, maternal ve paternal homologlar arasında DNA segmentlerihin birbirlerine çaprazlandığı (krosover) DNA parçasına karşılık gelen kiazma formasyonu içerir. Çoğunlukla her bir kromozom kolunda en az bir krossover olur, rekombinasyon dişideki mayozda erkekdeki mayozdan daha sık gerçekleşir. Sonuç olarak kromozomlar rastgele dağılırlar. Çünkü 23 kromozom vardır, burada kromozomların olası kombinasyonları 223 (> 8 milyon) olur. Rekomblnasyon esnasında oluşan genetik değişimlerle birlikte kromozortıal dağılım inanılmaz çeşitlilik ortaya çıkartır ve her gamet genetik olarak eşsizdir (tektir). Rekombinasyon oluşumu ve kromozomların bağımsız dağılımı, var olan bir genetik özellik ya da hastalıkla belli kromozom ■ bölgelerinin (ya da bağlı genler) kalıtımını ilişkileridirmede başvurulan bağlantı (îınkaj) analizinin yapılmasına bir temel oluşturur,
GEN EKSPRESYONUNUN DÜZENLENMESİ

Gen ekspresyonunu düzenleyen mekanizmalar genlerin fonksiyonunda kritik bir rol oynar. Fonksiyonel genomik denilen yeni alan, gen regülasyonu ve fonksiyonunu anlamanın fizyolojiyi daha iyi anlamaya ve yeni terapötilç fırsatlar sunmaya olanak sağlayacağı kavramına dayanmaktadır. Genlerin transkripsiyonu, öncelikle genlerin regülatör bölgelerindeki DNA dizilerihö bağlanan transkripsiyon faktörleri tarafından kontrol edilir. Aşağıda tarif edildiği gibi» transkripsiyon faktMerinde,- ki mutasyonlar beklenmedik kadar çok sayıda genetik bozukluklara ser bep olurlar. Gen ekspresyonu X-inaktivasyonu ve imprinting (iz sürme) gibi epigenetik (genetik dışı) olaylarla da etkilenir, burada ekspresyonun baskılanmasıyla ilişkili DNA metilasyonu olmuştur. Preder- Willi gibi (neonatal hipotoni, gelişim geriliği, şişmanlık, kısa boyluluk ve hipogonadizm) ve Albright kalıtsal osteodistrofi (paratlroit hormonuna direnç, kısa boyluluk, brakidaktili; bazı alt tipteki hormonlara karşı direnç) gibi bazı genetik bozukluklar genomik imprinting (iz sürme) sonucu ortaya çıkarlar. Gen ekspresyonu ile ilgili çoğu çalışmalar, transkripsiyonu kontrol eden genin regülatör DNA elemanları üzerinde yoğunlaşmıştır. Bununla birlikte, gen ekspresyonıınun hepsi aktif olarak regüle edilen mRNAoluşumu, protein translasyonu ve translasyon sonrası modifikasyonlar gibi bir seri basamaklardan oluştuğunu vurgulamak gerekir.
GENLERİN YAPISI
Bir genin ürünü çoğunlukla bir proteindir, fakat nadiren translasyonu yapılmayan RNA da olabilir. Genin neticede mRNA oluşturmak üzere bir araya gelmiş olan parçalarına ekzon denir. İntronlar ekzonlar arasında kalan, öncü RNA’dan olgun RNA oluşumu esnasında çıkarılan ara bölgeleri işaret eder. Gen lokusu, ekspresyonunu kontrol etmek için gerekli bölgeleri de gesinin (51 ön tarafındaki, dizilerdir, ön taraftaki regülatör bölgeler promoter olarak ta adlandırılırlar. En küçük promoter, TATA kutusu (bu TATA’ ya bağlanan proteini; TBP, bağlar ve aktif transkripsiyon kompleksinin oluşumunu sağlayan başlangıç dizilerinden oluşur. Transkripsiyonu sonlandırma sinyalleri, genin arka ya da 3’ kısmına yerleşmiştir. mF(NA’mn 3’ ucundaki AAUAAA dizisi gibi özel diziler, mRNA’nın sitoplazmaya taşınması, kararlılığı ve translasyonu etkileyen bir oluşum olan poliadenilasyon (Poly-A kuyruğu) bölgesini belirler. Regülatör bölge sınırlarının titiz bir testini yapmak, endojen genin normal gelişimsel, dokuya-özgün ve sinyale duyarlı özelliklerini söndürmek için yeterli olup olmadığını belirlemek için izole gen kuşatpı (flanking) DNA dizilerinin transgenik bir hayvanda ekspresyonunu gerektirir. Bu sadece birkaç gende başanlabilmiştir; in vivo olarak uzak regülatör dizilerin varlığına işaret eden geniş genomik dizilere ait normal gen ekspresyonunun sadece bir kısmını yerine getirebildiğini gösteren örnekler vardır. Bu yaklaşım, genleri regüle eden mekanizmaları anlamamız için kritiktir vebu yaklaşım ayrıca normal gen regulasyonun olmasını gerektiren gen tedavisi stratejileri için de uygundur.
Genler daha ayrıntılı olarak incelenebildikleri için DNA dizileri ve transkripsiyonu regüle eden transkripsiyon faktörlerinin sayılan orijinal olarak beklenilenden daha büyük olmuştur. Çoğu genler transkripsiyonun başlama bölgesinden itibaren 300 bp içinde 15-20 kadar farklı regülatör eleman içerirler. Bu yoğun olarak paketlenmiş olan promo- ter bölgesi, CAAT kutusu/enhancer (kuvvetlendirici) bağlama proteini (C/EBP), siklik AMP cevap elemanı bağlama proteini (CREB), seçici promoter faktörü 1 (Sp-1) ya da aktive edici protein 1 (AP-1) gibi her zaman bulunan transkripsiyon faktörlerinin bağlanma bölgelerini içerir. Bununla birlikte, hücreye özgü ekspresyonu içeren faktörler de bu dizilere bağlanabilir. Örneğin, başlıca heliks-loop -heliks (spiral-ilmik-spiral) (bHLH) proteinleri miyogenik genlerin promöterlerindeki E-kutusuna ve steroidogenik faktör l (SF-l) çoklu steroidogenik enzim genlerinin regülatör bölgesindeki özel tanıma bölgesine bağlanır. Anahtar regülatör elemanlar da proksimal promoterden biraz uzağa yerleşebilir, Globin ve immünogloblin genleri, örneğin, yapısal gen dizilerinden kilobazlarca uzaktaki loktıs kontrol bölgelerine sahiptir. Bu prornoterlere bağlanan özel transkripsiyon faktörü grupları ve enhancer dizileri transkripsiyonu regüle etmek için ortak bir kod geliştirmişlerdir. Bu durumda, bol bulunan faktörler daha az bulunan faktörlerle karşılıklı etkileşerek her genin özgün bir biçimde eksprese Olmasını ve regülasyonuniı sağlar: Aşağıda belirtildiği gibi, DNA’ya bağlanan transkripsiyon faktörleri gerçekte düzenleyici kontrolün sadece birinci basamağını oluşturur. Bü kompleksler asetilasyon ve fosforilasyon dahil çok sayıda sinyal yolu tarafından kontrol edilir. Sonunda, yeni gelen transkripsiyon faktörleri başlatıcı bölge ve TATA kutusunun etrafında toplanan bazal transkripsiyon kompleksinin bileşenleri ile karşılıklı etkileşime girer ve kararlı hale gelirler. Bü bazal transkripsiyon faktör kompleksi 30 dan fazla farklı proteinden oluşmuştur. Gen transkripsiyonu, RNA polimerazın DNA kalıbından RNA sentez etmeye başladığında gerçekleşir.
TRANSKRİPSİYONUN AKTİVE EDİLMESİ VE BASKILANMASI
Her gen, ister yerel veya zamansal ekspresyonunda, isterse ekstrasellüler uyarılara verilen cevabında olsun tamamen kendine özgün bir biçimde kontrol edilir. Transkripsiyon faktörlerinin eksprese olan genlerin hemen hemen %30 una hitap ettiği düşünülmektedir. Tanımlanmış genetik hastalıkların sayılarının artışı transkripsiyon faktörlerine bağlıdır. MODY (erişkinlik öncesi gençlik diyabeti) bozuklukları bu grup hastalıkların temsilcisidir; değişik adacıklardaki hücreye özgü transkripsiyon faktörlerinde görülen mutasyonlar değişik MODY tiplerinin oluşumuna sebep olur.
Transkripsiyonal aktivite üç ana mekanizmaya ayrılabilir:
-
Kromatin yapısını değiştiren olaylar, DNA’ ya transkripsiyon
faktörlerinin ulaşmasını arttırır. Örneğin, histon asetilasyonu kromatin yapısını açar ve bu transkripsiyon aktivasyonu ile ilişkilidir. -
Fosforilasyon gibi transkripsiyon faktörlerinin translasyon sonrası modifikasyonları, aktif transkripsiyon kompleksinin oluş- masını teşvik edebilir. Örneğin, CREB proteininin serin 133 üzerinden fosforilizasyonu konformayonel bir değişimi teşvik eder, bu da proteinler dahil pekçok transkripsiyon faktörünün aktivitelerini birleştiren bir faktör olan ÇREB-bağlama proteininin (CBP) devreye girmesini sağlar.
-
Transkripsiyonal aktivatörler represör bir proteinle yer değiştirebilir. Özellikle gelişme esnasında transkripsiyon faktörünün ekspresyon biçimi dinamik olarak değiştiğinden bu mekanizma çok sık görülür.
Doğal olarak, bu mekanizmalar birbirini dışlamaz ve çoğu genler bu olayların kombinasyonu ile aktive edilirler. Genel olarak transkripsiyonu baskılayan mekanizmalar bir anlamda transkripsiyonu aktive eden mekanizmalar kadar çalışılmamıştır. Bununla birlikte, gen ekspresyonunun baskılanması gen aktivasyonu kadar önemlidir. Bazı baskılama mekanizmaları aktivasyonun doğal sonucudur. Örneğin, baskılanma sık sık histon deasetilasyonu ve defosforilasyonu ile ilişkilendirilir. Nükleer hormon reseptörleri için transkripsiyonal sessizlik, histon deasetüaz aktivitesini içeren baskılama kompleksinin çalıştırılmasını gerektirir. Represör proteinin hatalı ekspresyonu bazen neoplazi ile ilişkilidir Premyelositik lösemide oluşan t(15;17) kromozom translokasyonu PML gçnini, retinoik asit a; reseptör (RAR ot) geninin bir parçası ile kaynaştırır Bu olay normal hücre farklılaşmasını dışlayacak bir şekilde düzenlenmemiş bir transkripsiyonal represyona sebep olur RAR ligantı (retinoik asit) ilave edilmesi reseptörü aktive eder, böylece represyon ortadan kalkar, hücreler faklılaşır ve sonunda apapitoz olur. Bu mekanizma tedavide önemlidir, çünkü retinoik asidin tedavi rejimine ilave edilmesi promyelositik anemili hastalarda yüksek oranda bir gerileme sağlar.
DNA’NIN KLONLANMASI VE DİZİ ANALİZİ
1970 lerin ortalarından beri sekiz Nobel ödülü, direkt yada indirekt olarak genetiğin kendi içindeki derinleşmeler kadar geniş ileri metodoloji alanındaki araştırmalara verilmiştir. Örnekler, revers transk- riptazın keşfi, restriksiyon enzimleri, plazmid klonlama vektörleri, DNA dizi analizi ve PCR’ı içermektedir. Rekombinant DNA tekniklerinin tarifi, DNA segmentlerinin manipülasyonu, analizleri ve ka- rakterize edilmesi için kullanılan yöntemler bü konunun kapsamı dışında kalmaktadır. Bu yöntemler genetik ve moleküler teşhislerde geniş çapta kullanıldıklarından DN A’nın klonlanması ve dizi analizi ile ilgili ana ilkeleri kısaca gözden geçirmek yararlı olacaktır.
GENLERİN KLONLANMASI
Klonlanma istenildiği kadar çoğaltılabilecek olan rekombinant DNA molekülünü yapmayı işaret eder. Genleri ve cDNA’lan klonlama yeteneği, bu ajanların yenilenebilen ve kalıcı olan bir kaynağının olmasını sağlar. Klonlama, DNA dizi analizi, nukleik asit hibridizasyon çalışmaları, rekombinant proteinlerin ekspresyonu ve diğer rekombinant DNA olayları için esastır.
DNA’ların klonlanması, klonlama vektörünün içine DNA parçasının girmesini ve takiben rekombinant DNA’mn konak hücre içinde çoğaltılmasını kapsar. Direkt klonlama stratejisi DNA fragmanının bakteri plasmidi içine girmesi şeklindedir. Plazmidler küçük, otomatik olarak replike olan, bakteri hücresi kromozomundan ayrı olarak çoğalan, çembersel DNA molekülüdür. DNA’nın eklenmesi olayı, DNA’ yı oldukça özel dizilerden kesen (çoğunlukla 4 ya da 6 baz çifti uzunluğunda) restriksiyon enzimlerinin kullanılması ile olur. Restriksiyon enzimleri, DNA fragmanlarının uçlarında,kendilerini plazmid vektörlere bağlayacak olaıı kömplementer, yapışkan uçlar oluştururlar. Çünkü plazmidler antibiyotiklere direnç genlerine sahiptirler, konak hücrede bu genlerin varlığı seçim ve DNA çoğaltımi (amplifikasyonu) için kullanılır.
Klonlama için şimdi çok değişik sayıda vektörler ve uygun konaklar kullanılmaktadır. Bunların çoğu küıüphane oluşturmak için kullanılmaktadır; kütüphane DNA klonlannın koleksiyonu demektir. Genomik kütüphane, genomik DNA’ dan türemiş olan bîr klo- ııu temsil eder. Örtüşen DNA fragmanları genomun tamamını temsil eder ve neticede lineer bir sıraya göre düzenlenebilir. Genomik kütüphaneler, Lamda (1) faj, kosm idler, bakteri yapay kromozomları ı.BACs) ve maya yapay kromozomları t YACs) gibi değişik . vektörler kullanılarak çoğaltılırlar. Faj kütüphaneleri çoğunlukla spesifik genlerin izole edilmesinde kullanılırlar. Kozmidler, BAC’lar ve YAC’Iar özellikle büyük genlerin çalışılmasında ve kromozom üzerinde genlerin sırasının belirlenmesi için yararlıdır. cDNA kütüphaneleri tipik olarak belli, bir dokudan kaynaklanan mRNA’dan türemiş klonlardır. Kalpten. elde edilen bir cDNA kütüphanesi özellikle kalp kası hücrelerinde sürekli olarak eksprese olan mRNA kopyalarını içerir. Bu nedenle, kalp cDNA kütüphaneleri kalbe özgü gen ürünlerince zenginleşecek ve karaciğer ve hipofit mRNA’tarınca "oluşturulan cDNA kütüphanelerinden de farklı olacaktır. Genomik kütüphanenin kompleksliğine örnek olarak, insan genomunun 3xl09bç olduğu ve genoma giren 1 fajin kütüphanesinde K^bç olduğunu düşünelim. Bu durmda, bütün genomik DNA yı göstermesi için en az 3x105 klona gerek vardır. Spesifik klonlar, DNA hibridizasyonu kullanılarak yüz binlerce klondan izole edilirler,
NUKLEİK ASİT HİBRİDİZASYONU
Nukleik asit hibridizasyonu, nukleik asitlerin, iki kömplementer ipliğinden birinin diğerine çok yüksek bir özgünlükle ağlanması ya da hibridizasyonu gerçeğinin avantajından dolayı moleküler biyolojinin temel prensibidir. Hibridizas- yonun amacı spesifik nukleik asit (DNA yada RNA) dizilerini diğer kompleks dizilerin içinde saptayabilmektir. Southern blot, Northern blot ve Kütüphane taramada bu teknikler kullanılır. Hibridizasyon tekniklerinin daha iyi adaptasyonu mikroarray DNA çiplerinin gelişmesine yol açmıştır.
Southern Blot Southern blot genlerin kayıp mı yoksa yeniden mi düzenlendiğini analiz etmek için kullanılır. Aynı zamanda restriksiyon fragman uzunluk polimorfızmini (RFLP) saptamak için de kullanılır. Genomik DNA restriksiyon endonükleazlarla sindirilir ve jel-elektro-forezi ile ayrılır. Bireysel fragmanlar daha sonra membrana transfer edilebilir ve spesifik radyoaktif DNA probları ile hibridize edildikten sonra belirlenebilir. Çünkü tek baz çiftinin yanlış eşleşmesi kısa DNA problarının (oligonukleotidler) hibridizasyonunu engelleyebilir, oligonukleotid spesifik hibridizasyon (OSH) denilen bir Southern blot varyasyonu kısa nukleotidler kullanılarak mutant genleri normallerden ayırmak için kullanılır.
HGF (insan genom projesi)’nin tamamlanmasıyla, bütün insan genleri klonlanıp dizi analizi yapılmış olacaktır. Sonuçta, bu klonlama olaylarının pek çoğuna gerek olmayacak ya da DNA dizileri ve markırlarını içeren geniş bilgi birikimi, bu işlemleri çok fazla kolaylaştıracaktır (aşağıya bakınız). Northern blot, farklı dokulardaki gen ekspresyonu düzeylerini ve biçimlerim analiz etmek için kullanılır. Northern blotta jel üzerinde mRNA ayrılır ve membrana aktarılır. Spesifik transkriptler radyoaktif işaretli DNA problan kullanılarak belirlenir. Bu teknik daha kapsamlı ve daha duyarlı olan revers transkriptaz (RT)-PCR ve DNA çipleri üzerine yerleştirilmiş gen ekspresyonu dizileri teknikleriyle çok çabuk yer değiştirmiştir.
Mikroarray Teknolojisi Genom çapındaki çalışmalara çok hızlı giren bir yaklaşım mikroarrayler ya da DNA çipleridir. Bu yaklaşımlar, ince cam ya da slikon yüzeylere milyonlarca sentetik nükleik asit dizilerinin sıralanmasını içerir. Floresanla işaretlenmiş DNA ya da RNA test örneği çiple hibridize olur ve bilgisayarlı tarayıcı (scanner) dizi eşleşmesini belirler. Mikroarrayler DNA dizi-terindeki varyasyonların belirlenmesini sağlar ve mutasyon analizleri ve genotiplemede kullanılır. Alternatif olarak, cDNA ya da genomik mikroarraylerle RNA örneklerini hibridleyerek büyük sayıdaki mRNA transkiptlerinin ekspresyon biçimi belirlenebilir. Bu yöntem fonksiyonel genetik çağında inanılmaz derecede büyük bir potansiyele sahiptir. Bir örnek olarak, mikroarrayler hastalığın sınıflaması, patofizyolojisi, prognozu ve tedavisinde yararlı bilgiler sağlayacak şekilde farklı lenfoma tilerinin genetik parmak izlerinin geliştirilmesinde kullanılabilir.
POLİMERAZ ZİNCİR REAKSİYONU
1985 te ortaya çıkan PCR, DNA analizlerinin gösterilmesinde devrim oluşturdu ve moleküler biyoloji ve genetik analizlerin bir köşetaşı haline geldi. Esasında PCR in vitroda spesifik DNA parçalarının klonlanmasmm (ampiifiye olması) hızlı bir şekilde yapılmasını sağlar. Verilen bir DNA dizisi için tasarlanmış PCR, primer kullanımı ile çok büyük bir özgünlük sağlar. Değişik döngülerden sonra DNA nın geometrik olarak çoğaltılması dikkate değer bir duyarlılık içinde gerçekleşir. Sonuç olarak, PCR tek bir hücre de dahil çok küçük örneklerden DNA’nın çoğaltılması için kullanılabilir. Bu özellikler kan hücresi, biyopsi materyali, otopsi ya da cerrahi örnekler, ya da tükrük ve saçtan gelen bir hücre de dahil değişik doku kaynaklarından DNAnın çoğaltılmasını sağlar. PCR, mRNA çalışmak içinde kullanılabilir. Bu durumda RT enzimi önce RNA’yı DNA’ya dönüştürmek için kullanılır, daha sonra PCR’la çoğaltılabilir. Daha çok RT-PCR olarak bilinen bu yöntem gen ekspresyonunun kantita- tif ölçümü için yararlıdır.
PCR, moleküler teşhis için bir anahtar eleman oluşturur. DNA dizi analizi dahil, çok sayıda dizileme teknikleriyle mutasyonları araştırmak için hızlı DNA (ya da mRNA) çoğaltılmasına ilişkin bir strateji sağlar. PCR aynı zamanda oldukça polimorfik iki veya üç nukleotidlik dizi tekrarlarının çoğaltılması için de kullanılır, bu da genetik bağlantı ya da ilgili çalışmalarda değişik polimorfik alicilerin takip edilmesini sağlar. PCR çeşitli mikrobik patojenlerin tanınmasında giderek daha çok kullanılmaktadır.
Mutasyon fonksiyonel sonuçlarına bakılmaksızın DNA’nın pirimer nukleotid dizisindeki herhangi bir değişiklik olarak tarif edilir. Bazı mutasyonlar zararlı, bazıları daha az zararlı olabilir ve bazılan evrimsel bir avantaj sunabilir. Mutasyonlar eşey (germ-line) hücrelerinde (sperm veya oositler) meydana gelebilir; bunlar gelecek kuşaklara aktarılabilir. Alternatif olarak, mutasyonlar embriyogenez esnasında veya somatik dokularda da meydana gelebilir. Gelişim esnasında ortaya çıkan mutasyonlar, değişik genetik yapılara sahip hücrelerin oluşturduğu dokularda görülen ve mozayiklik denen özel bir duruma yol açabilir. Eğer germline (eşey) hücrelerinde bir mozayiklik varsa, bir mutasyon bazı bireylere aktarıldığı halde bazılanna geçmez, bu durum bazen kalıtım şeklini tahminde kanşıklıklara yol açar. Yaşamsal önemi olmayan bazı somatik mutasyonlar; dokulardaki değişik fenotipik etkileri ile görülebilir (örneğin McCune-Albright sendromundaki pigmentli lezyonlar). Diğer bazı somatik mutasyonlar hücrelere büyüme avantajı sağlamalarından ötürü neoplazilerle ilişkilidir. Değişik DNAmetilas- yonu gibi epigenetik olaylar da gen ekspresyonunu etkileyebilir. Genişleme gösteren (aşağıya bakınız) triplet nukleotid tekrarları hariç, mutasyonlar genelde kararlıdır (stabildir).
Mutasyonlar yapısal olarak değişkendir- triploidilerde (fazladan bir kromozom takımı) tüm genomu içerebilir, veya kromozom ve genlerde çok büyük sayısal veya yapısal değişiklikleri içerebilir (Konu 66). Büyük delesyonlar, genin bir parçasını veya tamamını etkileyebilir veya çok sayıda gen içeriyorsa, bitişik gen sendromuna yol açabilir. Renk körlüğünde görüldüğü gibi, homolog genler arasındaki krosing- over eşit olmaz ise bileşik gen mutasyonu ile sonuçlanabilir. Tek nukleotidleri ilgilendiren mutasyonlara nokta mutasyonları denir. Bir pürin ile diğer bir pürinin (A<-tG) veya bir pirimidin ile diğer bir pirimidinin (C<->T) yer değiştirdiği mutasyonlara transisyon, bunun tersine (bir pürin bir pirimidin ile yer değiştirirse) transversiyon denir. Eğer bir DNA dizi değişikliği kodlama bölgesinde olur ve bir amino asitin yapısını değiştirirse buna miss ense (yanlış anlamlı) mutasyon denir. Proteinin farklı bölgelerinde amino asit değişimine neden olan bir missense mutasyon fonksiyonel sonuçlarına bağlı olarak belirgin fenotiplerin ortaya çıkmasına yol açabilir. Polimorfizmler en az %1 ’lik frekansa sahip dizi varyasyonlarıdır. Bunlar çoğunlukla fark edilebilir bir fenotiple sonuçlanmazlar. Bazılarının mRNA kararlılığını, translasyonu veya amino asit dizisini değiştirebilmesine karşın, genetik kodun dejenere oluşundan ötürü çoğunlukla tek baz çifti yerdeğişimleri protein kodlayan dizileri genelde değiştirmez. Bu tip sessiz baz yer değişimleri ve SNP’lere genetik testlerde çok sık rastlanır ve bunlar proteinlerin yapı ve işlevlerini değiştiren gerçek mutasyonlardan ayırt edilmelidir. Küçük nukleotid delesyonları veya insersiyonlar, kodon okuma çerçevesinde bir kaymaya neden olur. Çok yaygın olarak, okuma çerçevesi değişiklikleri, translasyonun sonlandırma kodonundan önce bitmesi ile oluşan farklı boylardaki anormal protein parçalan ile sonuçlanır (nonsense=anlamsız mutasyon). İntron dizilerinde veya ekzon kavşaklarındaki mutasyonlar, yeni kesim alanlan yaratır veya olanlan yok edebilir. Mutasyonlar genlerin regülatör bölgelerinde de olabilir, bunlar gen transkripsiyonunu azaltır.
Mutasyon Oranları Daha önce değinildiği gibi, mutasyonlar hastalıklar kadar genetik çeşitliliğin de önemli bir nedenini oluşturur. Çoğu mutasyonların sessiz oluşu ve testin fenotipik sonuçlan göstermek için yetersiz oluşu nedeniyle, insanda mutasyon oranlarını saptamak zordur. Mutasyon oranları genlere göre değişir fakat her hücre bölünmesinde yaklaşık 10-1 oranında meydana geldiği tahmin edilmektedir. Germline (eşey hücresi) mutasyonları (somatik mutasyonların aksine) genetik hastalıkların geçişine uygundur. Gelişim esnasında oosit populasyonunun çok erken evrelerde oluşmasından ötürü, oogenezin tamamlanması için yaklaşık 20 hücre bölünmesi gerekir, buna karşın puberte çağma ulaşıldığında spermatogeneziste yaklaşık 30 bölünme geçirilir ve ondan sonra da her yıl yaklaşık 20 hücre bölünmesi olur.
Netice olarak, yeni mutasyonları kazanma olasılığı dişi gametlere göre erkek gametlerde çok daha fazladır, bu durumda da anöploidi oranı artar. Böylece, spermatogoniumlardaki nokta mutasyonu insidansı baba yaşı ile birlikte artar (örneğin akondrodisplazi, Marfan sendromu, nöröfibramatoz). Her 10 spermden 1 tanesinin yeni bir zararlı mutasyon taşıdığı tahmin edilmektedir. Otozomal dominant ve X’e bağlı hastalıklar için mutasyon oranı hesaplamış olup bu oran her kuşakta lokus başına 10-5 kadardır, Çoğu monogenik hastalıkların nispeten nadir olmasından ötürü, yeni mutasyonlar olguların önemli bir parçası için geçerlidir. Bu genetik danışmanlık bakımından önemlidir, çünkü yeni bir mutasyon etkilenen bir bireye geçebilir fakat bu durum ebeveynlerin hastalığı diğer çocuklarına da geçirme riski altında oldukları anlamına gelmez. Gametlerin oluşumunun erken evrelerinde meydana gelerek gonadal musaizm’e yol açan mutasyonlar istisna teşkil etmektedir.
Eşit olmayan krosing over Normal olarak, germ hücrelerinde görülen DNA rekombinasyonu, değişen DNA parçalan arasındaki belli kavşak noktalannı korumak için dikkate değer derecede bir doğruluk gösterir. Bununla birlikte, homolog dizilerin yanlış eşleşmesi bir kromozomda gen delesyonu, diğerinde gen duplikasyonu olacak şekilde eşit olmayan bir krosing övere neden olur. Örneğin, büyüme hormonu (GH) gen delesyonlannın önemli bir bölümü eşit olmayan krosing-overden ileri gelir. GH geni, bir büyüme hormonu varyant geni, yapısal olarak benzerlik gösteren çeşitli koriyonik somatomammotropin genleri ve psödogenleri (oldukça homolog fakat fonksiyonel olarak normal bir genin inaktif akrabaları) içeren büyük bir gen ailesinin üyesidir. Bu gibi gen aileleri ardışık şekilde düzenlenmiş (tandem) çeşitli homolog DNA dizileri taşıdığından, rekombinasyon yapmaya ve sonuçta da gen delesyonu ve duplikasyonuna meyillidir. Diğer taraftan eşit olmayan krossing over sonucu olan PM22 geninin duplikasyonu, gen dozajt artışına ve tip IA Charcot-Marie-Tooth hastalığına sebep olur (Konu 379). Eşit olmayan krossingoverin neden olduğu PMP22 delesyonu, basınç felcine, kalıtsal hassasiyet denen belli bir nöropatiye neden olur.
Glukokortikoid ile iyileştirilebilen aldosteronizm (GRA) normalde kromozom 8q üzerinde ardışık yer alan aldosteron sentazı kodlayan geni (CYPIIB2) ile steroid 1 ljî-hidroksilaz geni (CYPIIBI) arasındaki yeniden düzenlemeden ileri gelir. Bu iki gen %95 identik olup eşit olmayan krossing-over ile gen duplikasyonu veya delesyonu yapmaya yatkındır. Krosing-over ile yeniden düzenlenen genin ürünü, aldosteron sentazın kodlama dizisine bitişmiş olan liP-hidroksilazın regülatör bölgesini içerir. Netice olarak, bu bitişik genin ürünü olan enzim, adrenal bezin adrenokortikotropik hormona (ACTH)-bağlı zonunda eksprese olur ve mineralokortikoidlerin aşın salınmasına ve hipertansiyona neden olur.
Gen çevrilmesi homolog genetik bilginin karşılıklı olmayan değişimi (non-resiprokal krosing-over) anlamina gelir; bu durum çoğu kez sanıldığından büyük olasılıkla daha yaygındır. İnsan genetiğinde gen çevrimi, bir genin içindeki bir bölümün diğer allel veya lokusdan kopyalanan homolog bir segmentle nasıl yer değiştirildiğini açıklamak için kullanılır; bu genetik değişiklikler birkaç nukleotidten birkaç bin nukleotide kadar olabilir. Gen çevrilmesi sonucunda, ebeveynlerde ayrı diziler halinde olan iki kromozoma ait kısa DNA parçasının idendik hale gelmesi mümkün olur. Bu olayın pratik sonucu, genellikle genin fonksiyonunu değiştirecek şekilde akraba genler arasında gerçekleşen gen çevrilmesi esnasında nukleotid yer değişiminin meydana gelmesidir. Hastalık durumlarındaki gen çevrilmesinde, bir gen ile akraba bir psödogen arasında genler arası DNA değişimi olur.
Örneğin 21-hidroksilaz geni (CYP21 A) fonksiyonel olmayan bir psödogenin yanında bulunur. Konjenital hiperplazili hastalardaki CYP21A geninde görülen pek çok nukleotid yerdeğişimi, psödogenlerdeki dizilere karşılık gelmektedir ki bu durum da gen çevrilmesinin bir mutagenez mekanizması olduğunu düşündürmektedir.
BASİT MENDEL KALITIM MODELLERİNİN DIŞINDAKİLER
Mitokondrial Hastalıklar Her mitokondrion değişik sayıda halkasal kromozom içerir. Mitokondrial DNA esas olarak oksidatif fosforilasyon ve ATP üretimine katılan solunum zinciri elemanları olan transfer RNA ve proteinleri kodlar. Sperm zigota önemli bir sitoplazmik içerik katkısında bulunmadığı için mitokondrial genom anne yolu ile kalıtılır. Tüm çocuklar etkilenmiş anneden hastalığı alır fakat bu hastalık etkilenmiş babadan çocuklarına aktanlmayacaktır. Hücre bölünmesi esnasında, yabanıl tip ve mutant mitokondri oranı hücreden hücreye değişir; kusurlu mito- kondri gruplarındaki farklılıklara heteroplazmi denir ve bu durum mitokondrial hastalıklarda çok görülen fenotipik çeşitliliği kısmen açıklamaktadır.
Mozaisizm (=mozayiklik) Bir bireyin dokularında iki veya daha fazla genetik olarak farklı hücre hatlarının bulunmasına mozaisizm denir. Mozayiklik embriyonik, fötal veya uterus dışındaki gelişim esnasında meydana gelen mutasyonlardan ileri gelir. Mutas- yonun ortaya çıktığı gelişim evresi, eşey hücre hattı ve/veya somatik hücrelerin olaya katılıp katılmayacağını belirler. Turner send- romlu hastalarda ortaya çıktığı gibi birden fazla hücre hattının sürdürülmesine yol açan kromozomal mozasizm, embriyonik gelişimin başlangıcındaki mitotik bölünmeler esnasındaki kromozom ayrılamamasından ileri gelir. Somatik mozasizm genetik olarak değişmiş hücrelerin parçalı dağılımı ile karakterizedir. Örneğin McCune-Albright sendromuna, gelişimin erken evrelerinde görülen uyarıcı Ga proteinindeki (Gscc) aktive edici mutasyonlar neden olur (Konu 343). Klinik fenotip mutasyonun doku dağılımına bağlı olarak değişir; belirtiler, cinsiyet steroidlerinin salınmasına ve erken puberteye neden olan ovaryum kistleri, polyostatik fib- röz displazi, “cafe-au-lait” deri pigmentasyonu, hipofiz adenomla- rından büyüme hormonu salgılanması, aşın salgı yapan otonom tiroid ııodüllerini içerir.
X-inaktivasyonu, Imprinting ve Uniparental Dizomi Geleneksel Mendel kurallarına göre mutant genin parental oriini ile fenotipin etkinlik derecesi arasında ilişki yoktur. Bununla birlikte, bu kuralın önemli istisnaları vardır. X-inaktivasyonu dişilerin her hücresinde iki X kromozomdan biri üzerindeki tüm genlerin etkinliğini önler. Gen inaktivasyonu otozomların seçilmiş bazı kromozom alanlarında da meydana gelir. Genomik imprinting denen bu olay, bir allelin parental orijinine göre tercihli etkinlik göstermesine yol açar. Genomik impriting, hastalığın geçişi bunu sağlayan ebeveynin cinsiyetine bağlı olduğu hastalıklarda fizyopatolojik bir öneme sahiptir ve böylece belli genetik hastalıkların etkisini göstermesinde önemli rol oynar. İki klasik örneği Prader-Willi Sendromu ve Angelman sendromudur.
Prader-Willi sendromu azalmış fetal aktivite, obesite, hipotoni, zihinsel gerilik, kısa boyluluk ve hipogonodotropik hipogonadizm ile kakarterizedir. Prader-Willi sendro- munda delesyonlar sadece paternal 15. kromozomda meydana ge- ler. Bunun aksine, Angelman sendromlu hastalar, zihinsel gerilik, tutulumlar, ataksiler ve hipotoni ile karakterize olup 15. kromozomun aynı bölgesinde delasyonlar görülür; bununla birlikte bu delesyonlar maternal 15. kromozom üzerinde yer alır. Bu iki sendrom uniparental dizomiden de ileri gelebilir. Bu durumda sendromlar, 15. kromozom üzerindeki delesyonlardan değil her iki parental kromozomun alınması (Prader-Willi sendromu) veya her iki maternal kromozomun alınmasından (Angelman sendromu) ileri gelebilir.
Özgün doku ve bireysel hürcelerde paternal ve maternal alicilerden gelen mRNA ekspresyon seviyelerini incelemek çok zor olduğundan, imprinting (=genetik iz sürme) ve allelik dışlama ile ilgili olaylar belki de şu anda bilinenden çok daha yaygın olabilir. Genetik impriting veya uniparental dizomi diğer birkaç hastalık ve kötü huylu tümör olgularının patojenizine katılır. Hida- tidiform benler normal sayıda diploid kromozom içerir fakat hepsi paternal orijinlidir. Bunun tersi bir durum 46 maternal orijinli kromozom içeren ovaryum teratomlarında görülür. Kansere yatkınlığa yol açan Backwith-Wiedemann sendromunun (BWS) patogenezinde insulin-benzeri büyüme faktörü II(IGF-II) için genetik iz süren bir genin etkinliği söz konusudur. Bu çocuklar orga- nomegali ve hemihipertrofili somatik çıkıntılar gösterir ve bunlarda Wilm Tümörü gibi embriyonal tümörler riskinde artış vardır. Normal olarak IGF-II geninin sadece babadan gelen kopyesi aktif, anneden gelen kopyesi inaktiftir. IGF-II’nin genetik iz sürümü, proteine çevrilmeyen bir RNA kodlayan H19 geni ile düzenlenir. H19 metilasyonundaki bir hata veya metilasyonun yokluğu IGF-II genetik iz sürümünde bir rahatlamaya ve her iki allelin de etkilendiğini göstermesine neden olur. DNA dizi değişiklikleri ile ilişkili olmayan gen etkinliğindeki kalıtımla geçebilen değişiklikler epige- netik etkiler olarak bilinir; bu değişikliklerin insan hastalıklarında ve belki de yaşlanmada rol oynadıkları giderek daha çok ortaya çıkmaktadır.
Somatik Mutasyonlar Kanser sendromlannın çoğunda, tümör oluşumuna kalıtsal bir yatkınlık vardır. Bununla birlikte, neop- lastik süreç ilave somatik mutasyonlann olmasını gerektirir (Konu 81). Retinoblastomada, retinoblastoma geninin (RB) her iki kopyası iki somatik olayda (sporadik retinoblastoma) inaktive olduğu zaman veya diğer allelinde kalıtsal bir kusur olan kişide normal allel somatik kayıba uğradığı zaman (kalıtsal retinoblastoma) tümör gelişir. Bu ”iki-vurgunlu” model MEN-1 (Konu 339) ve norofibroma- toz tip 2 gibi diğer kalıtsal kanser sendromlarına da uyar. Kusurlu allel, etkilenen dokudaki tümör supresör geninin resesif kaybı ile ortaya çıkan tümörleşme yolu ile dominant bir şekilde aktanlır. Diğer durumlarda, kanser gelişimi tipik olarak pek çok gende somatik kusur olmasını gerektirir, bu sürece çok-basamaklı karsinogenez denir.
Nukleotid Tekrar Genişlemesi Hastalıkları
Bir kısım hastalıklar belli bir eşik seviyesinin üstündeki trinukleotid tekrar sayısındaki artışla ilişkilidir. Artışlar bazen Huntington hastalığında veya spinal ve bulbar atrofinin X’e bağlı formunda (SBMA, Kenedy sendromu) olduğu gibi genin kodlama bölgesinde yer alabilir. Diğer durumlarda artışlar büyük olasılıkla genin regülatör dizilerini değiştirir. Bir genişleme varsa, bu DNA parçası kararlı değildir ve hücre bölünmesinde daha da genişlemeye meyillidir. Nukleotid tekrarının uzunluğu genellikle hastalığın şiddeti ile eşgüdümlüdür. Tekrar uzunluğu bir generasyondan diğerine artış gösterirse, belirtiler kötüye gidebilir veya daha erken yaşlarda gözlenebilir hale gelir; bu olaya “antisipasyon”= önceden tahmin denir. Örneğin Huntington hastalığında hastalığın başlama yaşı ile triplet kodonunun genişleme uzunluğu arasında biş parelellik vardır. Önceden tahmin, trinukleotid tekrarlarındaki dinamik mutasyonlarla ortaya çıkan diğer hastalıklarda da gösterilmiştir. Tekrar sayısı dokuya-özgün şekilde de değişebilir. Myotonik distrofide, CTG tekrarı kas dokusunda lenfositlerdekin- den on kat daha büyük olabilir.
Allel frekansları etnik gruplar ve coğrafık bölgeler arasında değişir. Örneğin CFTR genindeki heterozigot mutasyonlar, Avrupa orijinli populâsyonlarda oldukça yaygın bir şekilde görülür, fakat Afrika populasyonlannda nadirdir. Bazı allel varyantlarının selektif avantaj sağlaması nedeniyle allel frekansları değişebilir. Örneğin, özellikle Batı Afrika’da yaygın olarak görülen orak-hücresi mutasyonlan bakımından heterozigot bireyler, eritrositlerin Plasmodium parazitlerine daha uygun koşullar sağlamaları yüzünden sıtma bulaşma daha dirençlidir. Orak hücre geni bakımından homozigöt olanların şiddetli anemi ve orak hücresi krizleri geçirmelerine karşın , heterozigot olanlar sıtmadan dolayı azalmış bir morbidite ve mortalite göstererek daha yüksek bir yaşama şansına sahip olur; bu olay mutant allelin frekansında artışa yol açar. Daha kısıtlı bir gen havuzları olması nedeniyle resesif koşullar coğrafik olarak ayrılmış popülasyonlarda daha egemendir.
Atletik İlişki ve Bağlantı Sapması İnşamla hastalıkların onaya çıkmasına veya hastalıklara hassasiyetin artmasına neden olan genlerin haritalanması için iki primer strateji vardır; (1) bilinen bir genetik kalıtım modeline (yukarıya bakınız) dayanarak veya genetik model bilinmiyorsa etkilenen akraba çiftleri inceleyerek klasik bağlantı kurulabilir; veya (2) bu genler atletik ilişki çalışmaları ile haritalanabilir. Belli bir hastalıkta bir allelin frekansının önemli ölçüde artma veya azalmasına allelik ilişki (“allelic association”) denir. Bağlantı ve ilişki çeşitli bakımlardan farklılık gösterir. Genetik bağlantı ailelerde veya kardeşler arasında gösterilebilir. Diğer taraftan, ilişki incelemeleri, etkilenen bireylerin popu- lasyonu ile normal bir populasyonu karşılaştırır, İlişki incelemeleri. akraba olmayan etkilenmiş bireylerle karşılaştırılan kontrolleri içeren oİgu-çalışmaları veya etkilenen çocuklara geçen veya geçmeyen ailelerin frekansını karşılaştıran aileye-dayalı çalışmalar şeklinde olabilir.

Allelik ilişki çalışmaları, özellikle kompleks hastalardaki hassas genlerin tanınmasında yararlıdır. İki lokustaki alleller beklenildiğinden daha sık (bilinen allel frekansları ve rekombinasyon oranlarına dayanarak) birlikte oldukları zaman, bunların bağlantı sapması içinde olduklarından söz edilir. Z mutasyonu normal allelin Y olduğu, bir hassasiyet lokusunda meydana gel- miştir. Mutasyon, A ve B allellerini içeren polimorfık böglenin yar kmındadır. Zamanla, A ve Z allellerini taşıyan kromozomlar birikir ve populasyondaki kromozomların %10’unu temsil eder. Hastalık hassasiyet geni Z’nin tercihan veya sadece A alleli ile birlikte olması bağlantı sapmasını gösterir. A allelini taşıyan tüm kromozomların hastalık genini taşımamasına karşın A alleli, Z geni ile olan olası ilişkisinden ötürü artan bir riskle karşı karşıyadır. Bu model, hastalığa hassasiyeti daha kesin tahmin etmek için, gelecekte Z genini doğrudan tanımanın mümkün olabileceğine işaret etmektedir.
POLİGENİK HASTALIK VE KOMPLEKS GENETİK ÖZELLİKLER
Poligenik ve Multifaktöriyel Hastalıklara Yaklaşım Kardiyovasküler hastalık, hipertansiyon, diyabet, astan, psikiyatrik bozukluklar ve belli kanserler gibi pek çok yaygın hastalığın etkinlik derecesi, genetik, altyapı, çevresel koşullar ve yaşam tarzı ile belirlenir. Fenotipe çok sayıda genin katıldığı düşünülürse bu özelliğe poligenik denir veya çok sayıdaki genin çevresel koşullarla etkileşim içinde olduğu kabul edilirse bu özelliğe multifaktöriyel denir. Kompleks özelliklere ait genetik modeller, genetik heterojenite ve diğer genlerle ve çevre ile etkileşimlere açık olmalıdır. Kompleks genetik özellikler özelliğin patogenezini sağlayan asıl genlere bağlı olmayan değiştirici genler tarafından etkilenebilir. Bu tip gen-gen etkileşimi, veya epistazis; multipl genlerde patolojik bir ienutip-ortaya çıkarmak için çök sayıdaki varyasyonların beraberce bulunmasını gerektiren poligenik özelliklerde önemli bir rol oynar. Gen-çevre etkileşimleri, çoğu monogenik ve poligenik hastalıklarla ilişkilidir. Fenilketonuride, hastalığın etkinlik derecesi sadece fenilalanin genindeki mutasyonlara değil aynı zamanda alınan fenilalanin aminoasit miktarına da bağlıdır.
KROMOZOM ANOMALİLERİNİN TİPLERİ VE İNSİDANSI
Mayozda ya da erken evrelerdeki bölünmelerdeki hatalar oldukça yüksek frekansta olur. Örneğin, bütün gebeliklerin en azından % 10 ila 25 i arasını anormal kromozomlu gebelikler oluşturur. Bunların büyük bir oranı gebeliğin çok erken evrelerinde sonlanır, Bununla birlikte, klinik olarak tanınan gebelikler arasında bile yaklaşık olarak fötusun %10’u kromozom bakımından dengesizdir. Farklı tipteki kromozom anomalilerinin oluşumu, klinik olarak tanınan üç tip gebelik için; kendiliğinden düşükler, ölü doğumlar ve canlı doğumlar, Tablo 66-2 de özetlenmiştir. En çok rastlanan anomaliler fötusta ilave (trizomi) veya eksik (monozomi) bir kromozom veya ilave bir (triploidi) veya iki (tetraploidi) kromozom setini içeren sayısal anomalilerdir. En önemli klinik kromozom bozukluklarının çoğu yapısal yeniden düzenlenmeleri içerdiği halde yapısal kromozom anomalileri daha az görülür.
Oldukça sık rastlanılan anomali, kendiliğinden olan düşüklerin yaklaşık %25 inde ve yeni doğanların %0.3 ünde gözlene trizomi- dir. Bütün kromozomlardaki trizomiler şimdi embriyoda ya da fötusta belirlenmektedir, fakat değişik kromozomlardaki frekansı dikkate değer derecede farklılık gösterir. Örneğin, trizomi 16 olan- ğanüstü sık görülüp spontan düşüklerdeki trizomilerin 1/3 ünün sebebi olurken, trizomi 1, 5, 11 ve 19 daha az sıklıkta görülmektedir. Elde bulunan deliller bu çeşitliliğe iki sebep göstermektedirler; (1) bazı kromozomlar (örn, kromozom 16) mayozda diğerlerine göre daha çok anormal kromozom ayrılması veya nondisjunction gösterirler; ve (2) gelişme için potansiyel, farklı trizomik koşullara göre değişir, bazıları gebeliğin erken evrelerinde ortadan kaldırılabilir diğerleri gebeliğin klinik olarak tanınmasına kadar yaşarlar ve bazıları da (örn. trizomi 13, 18, 21 ve seks kromozomu trizomileri) doğuma kadar yaşayabilirler.
KROMOZOM SENDROMLARI
Çoğu kromozom anomalili gebelikler daha uterusta iken sona erdikleri halde bazı durumlarda doğuma kadar devam edebilirler. Bunların en iyi belirlenenleri bir kromozomun kaybolması veya kazanılması ile dengesiz translokasyonlar sonucu oluşan anomalileri de içeren sayısal anomalilerdir. FISH ve diğer moleküler çalışmalar, mikrode- lesyon sendromları ve iz-sürme (imprinting) sendromları olarak da bilinen iki yeni kromozom anomalisinin belirlenmesini sağlamıştır.
Sayısal Anomaliler Hemen hemen tüm sayısal anomali tiplerinin hepsi prenatal olarak ortadan kaldırılır, böylece canlı doğanlar arasında sadece küçük olan ve az gen taşıyan otozomlar veya seks kromozomlarına ait anomalileri taşıyanlar belli bir sıklıkta bulunur. Klinik olarak bunların en önemlisi en çok Down sendromuna neden olan trizomi 21 ’dir. Populasyonun anne yaşı yapısına dayanarak ve prenatal tetkiklerin kullanılmasıyla trizomi 21 in insidansı 1/600- 1/1000 olarak belirlenmekte, bu da onu canlı doğan bireyler arasında en sık rastlanan kromozom anomalisi yapmaktadır. Çoğu trizomiler gibi, trizomi 21 insidansı anne yaşı ile oldukça ilişkilidir, 20 yaşındaki kadınlar için oran yaklaşık 1/1500 canlı doğum iken, 45 ve üzeri yaştaki kadınlarda bu oran 1/30’a kadar yükselmektedir.
Trizomi 21 ’e ilave olarak, canlı doğumlarda sadece diğer iki otozomal trizomi olan trizomi 13 ve 18 belli bir sıklıkta görülür. Canlı doğumlarda trizomi 13 ve trizomi 18 için insidansı sırası ile 1/20.000 ve 1/10.000 dir. Yaşam süresi beklentisi normale yakın olan trizomi 2’in aksine, trizomi 13 ve 18’in her ikisi de tipik olarak yaşamm birinci yılında ölürler.
Üç seks kromozomu trizomisine - 47,XXX, 47,XXY (Klinefelter sendromu) ve 47,XYY- yenidoğanlarda yaklaşık 1/2000 oranında olacak şekilde oldukça sık rastlanılır. Bütün trizomilerden bu üçü çok az fenotipik komplikasyonlar gösterir. Esasen, klinefeiter sendromunda kısırlık hariç (Konu 335) öyle görünüyorki bu gibi trizomisi olanların çoğu belirlenemeyecektir. 47,XYY durumundaki fazlalık kromozom küçüktür ve çok az gen taşımaktadır. Yye bağlı genlerin çoğu testiküler gelişim ve spermatogenez ile ilgilidir. Böylece Y-ye bağlı genlerin dozaj dengesizliği, diğer gelişim işlemlerinde oransal olarak çok az etki yapar. 47,XYY genotipi uzun boyla ilişkilidir. Önceden varsayıldığı gibi cezalı populasyonlarda bunun prevalansının artmasının antisosyal davrasnışta rolü olduğu açık değildir.
47,XXX ve 47,XXY koşullarında durum farklıdır- X kromozomu çoğu normal gelişim için gerekli olan 1000 den fazla gen içermektedir. O halde 47,XXX ve 47,XXY olan bireyler dozaj dengesizliğinin katastrofik sonuçlardan nasıl kurtulur? Yanıt X kromozomu gen ekspresyonunun biyolojisinde yatmaktadır. Normal dişilerde, somatik hücrelerde X kromozomunun biri inaktive olmuştur. Paternal veya maternal X kromozomu inaktivasyonu her bir somatik hücrede rastlantısal olarak olur ve böylece X-e bağlı genlerin çoğunun erkek ve dişilerde eşit eksprese olmasını sağlar ve dozaj telafisi mekanizması olarak iş görür. İnaktivasyon olayı gelişmenin blastosist evresinde meydana gelir; bundan önce her iki X kromozomu aktifidir. İlaveten, germ hücreleri için inaktivasyon kuralları somatik hücreler için olandan farklıdır, dişi germ hücrelerinde her iki X kromozomu aktif kalır, halbuki erkek germ hücrlerinde X kromozomu inaktiftir.
Ayrıca X-e bağlı genlerin hepsi inaktif değildir. X kromozomunda bulunan genlerin bazıları inaktivasyon mekanizmasından kaçar ve her iki X kromozomundan ekprese olurlar. Klinefelter sendromu gibi bozukluklarda, bazı genler fenotipik anomalilerle sonuçlanacak şekilde her iki X kromozomundan eksprese olabilirler. Klinefelter sendromlu bireyler küçük testislere, hiyalinize seminifer tübüllerine ve azospermi ve ya ağır oligospermi- ye sahiptir. Testosteron düzeyleri değişken olarak azalmıştır ve genelde jinekomasti ve hadımsı (eunuchoidal) vücut oranlarına sahiptirler. Bazı bireylerde antisosyal davranış ve hafif zihinsel kusurlar görülür. 47,XXX genotipli dişiler büyük olasılıkla hafif zihinsel kusurlu olurlar ve kısmi kısır olabilirler. Bu özelliklere rağmen, seks kromozomu trizomileri otozomal kromozom anöploidileri ile karşılaştırdığında nispeten küçük fenotipik komplikasyonlara sebep olurlar. Kural olarak monozomik olanlar fötal evrede ölürler ve sonuçta otozomal monozomiler sadece spontan düşüklerde çok seyrek
Yapısal Kromozom Anomalileri Yapısal yeniden düzenlenmeler kromozomların kırılma ve yeniden birleşmelerini kapsar. Sayısal anomalilerden daha az oldukları halde, genetik danışma açısından ayrı bir mücadele konusu ortaya çıkarırlar. Çünkü yapısal anomalilerin sayısal anomalilerden farklı olarak, klinikte”dengeli’’ formları normal kişilerde bulunabilir fakat gelecek kuşağa kromozom anomalisinin kalıtsal bir formu olarak ’’dengesiz” bir biçimde aktarılabilir.
Yeniden düzenlenmeler farklı kromozomlar arasında karşılıklı materyal değişimini (translokasyonlar) veya tek bir kromozomdaki kayıp, kazanma veya yeniden düzenlemeleri (örn. delesyonlar, duplikasyonlar, itıversiyonlar, halka veya izokromozonılar) içerebilir. Özel klinik öneme sahip olan translokasyonlar temel olarak iki tahmin edilir; bu spontan düşüklerin büyük çoğunluğu sadece trizomi 13, 18 ve 21 lerdlr ve seks kromozomu trizomilerinin de doğuma kadar yaşayanları odukça çoktur.
Robertson ve resiprokal. Robertson yeniden düzenlenmeleri, iki akrosentrik kromozomun (kromozom 13, 14, 15, 21 ve 22) uzun kollarının iki kromozomun orijinal genetik materyalini içerecek şekilde bir araya gelmesiyle kaynaşmış bir kromozom oluşturması şeklinde oluşan özel bir translokasyon sınıfıdır. Eğer Robertson translokasyonu dengesiz formda ise monozomik ve trizomik gebelik olur. Örneğin, Down sendromu vakalarının yaklaşık %3 ü ekseriya 14. ve 21. kromozomlarda görülen dengesiz Robert son tipi translokasyonlara atfedilebilir. Bu durumda, etkilenmiş olan birey bir yapısal olarak normal kromozom 14, 2 yapısal olarak normal kromozom 21 ve bir kaynaşmış kromozom 14/21 olmak üzere 46 kromozoma sahiptir. Bu etki kromozom 14 ün normal dip- loit dozajına ve kromozom 21 in Down sendromu ile sonuçlanan üç katı dozajına sebeb olur. Benzer şekilde, trizomi 13 sendromlu bireylerin küçük bir oranı dengesiz Robertson translokasyonundan dolayı klinik olarak etkilenmiştir.
Resiprokal translokasyonlar tki kromozom arasındaki karşılıklı değişimdir. Bu durumda, dengesiz translokasyonlarla ilgili feno- tipik sonuçlar iki kromozom arasında değiştirilmiş olan materyalin miktarını belirleyecek olan kırılma noktalarının yerine bağlıdır. Çoğu resiprokal translokasyonlar özgün kırılma noktası setleri içerdiklerinden, herhangi bir durumda fenotipik sonuçları önceden belirlemek zordur. Genel olarak şiddeti, dengesiz translokasyonlu bireylerde yok olan ya da fazla olan kromozom materyalinin miktarıyla belirlenir.
Kromozomlar arasındaki yeniden düzenlemelerle birlikte, kromozom içinde görülen yapısal anomalilere ait pek çok örnek vardır. Bunların en sık ve zararlı olanı, delesyonlarla oluşan kromozom materyalinin kaybıdır. Çok iyi karaterize edilmiş iki delesyon sendromu, Wolf-Hirschhorn sendromu ve Cri-du-chat sendromu olup, kromozom 4p ve 5p deki nispeten küçük kromozom parçalarının kaybolması sonucu ortaya çıkarlar. Bununla birlikte, her biri çoklu doğumsal anomaliler, gelişme geriliği, şiddetli gerilik ve kısa ömür uzunluğu ile ilişkilidir.