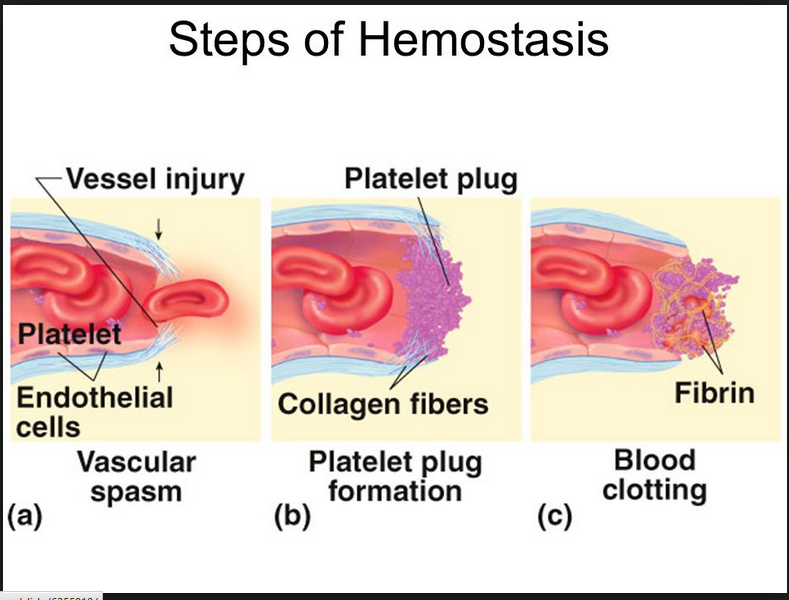
Normal hemostaz, sıvı kan akışını ve damar yatağının yapısal bütünlüğünü sürdüren prokoagülan ve antikoagülan faktörlerin fizyolojik dengesini içerir. Vasküler hasar, kan kaybını önlemek için trombositlerin toplanması ya da fibrin plağı üretilmesini amaçlayan pıhtılaşmanın başlamasına neden olur. Bu işlemi pıhtının sınırlanması, yara iyileşmesi, pıhtının çözünmesi, yeniden doku üretimi ve yeniden şekillenme aşamaları takip eder. Sağlıklı kişilerde bu reaksiyonların hepsi devamlı ve dengeli bir biçimde ortaya çıkar, örneğin; kanama durdurulur, ancak kan damarları açık kalır ve organlara yeterli miktarda kan gönderilir. Bu aşamalardan bir veya birkaçı, kalıtımsal eksiklikler veya edinilmiş anomalilerden dolayı bozulduğunda oluşan düzensiz hemostaz kanama veya tromboembolik komplikasyonlarla sonuçlanabilir. Arterial ve venöz sistemlerdeki kan akışı birbirinden çok farklıdır ve koagülasyon sistemine farklı gereklilikler yükler. Basınçlı arterlerde, göreceli olarak küçük vasküler hasarlar hızla büyük kan kaybına neden olabilir. Bunun için arterlerdeki koagülan yanıtlar kan kaybını hızlıca durdurabilecek kapasitede olmalıdır. Trombositler bu yanıtta çok önemlidirier, hemen kan kaybını durdururlar ve daha sonra hemostazın en sonunda oluşan fibrin şekillenmesini lokalize eden ve hızlandıran olaylar için aktif bir yüzey hazırlanmasını sağlarlar. Venöz dolaşımda tersine daha düşük kan akışı daha yavaş kanamaya neden olur. Burada trombositlerin görevi daha az önemlidir, venöz hemostazın dengesini kontrol eden kilit reaksiyon trombin oluşma hızıdır. Kullanılan antikoagülan ajanlarla da bu farklılıkların altı çizilir; aspirin gibi antiplatelet ajanlar koroner arter trombüsünu önlemek için, heparin ve warfarin gibi antitrombinesaslı uygulamalar ise derin-ven trombozuna karşı koruyucu amaçlı kullanılır.
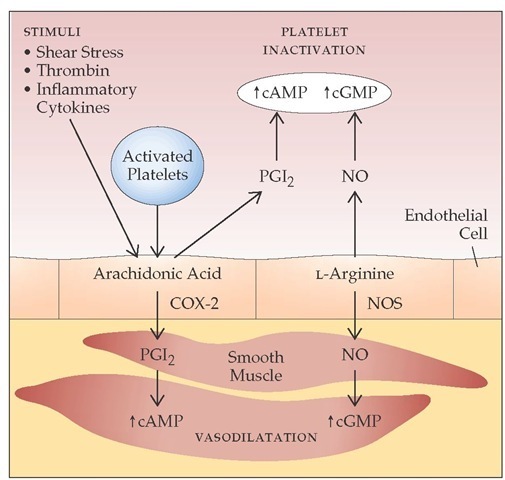
Damar endotel hücreleri (ECier), bir bariyer olarak fonksiyon görür ve kanın yüksek trombojenik subendotelyal içerikler ile temas etmesini engeller. Aslında; aterosklerotik lezyonların gelişiminin erken safhasında endotelyal disfonksiyonun önemJi olduğu görülmektedir. Normal intakt ECler güçlü antikoagülan fonksiyonlara sahiptir ve prostasiklin, nitrit oksit, adenosin difosfataz (ADPaz) ve plasminojen aktivatörü salgılar. Prostasiklin ve nitrik oksit trombozu önlemede ikili mekan izmaya sahiptir. Prostosiklin ve nitrik oksitin ikisi de düz kas hücrelerinde vazodilasyona neden olur ve böylece kan akışı artar ve damar çeperi ile trombosit teması en aza indirilir. Prostasiklin ve nitrik oksit damar endotel hücrelerinden aynı zamanda kan dolaşımına da salgılanır, trombositler içinde döngüsel adenosin monofosfat (cAMP) üretimini arttırır ve bu da trombosit aktivasyonunu ve trombosit kümeleşmesini baskılar. Ancak; ECler hasarlandığında veya aktive olduğunda koagülan özelliklerin dengesi hızla prokoagülan duruma kayar. Bu fonksiyon, hem EClerin kendisi tarafından hem de vasküler hasar sonucunda ortaya çıkan subendotelyal matriks tarafından oluşturulur. Aktive olmuş ECler yüzeylerinde yapışkan ligand olan E ve P selektinleri,
integrini, trombosit-EC adezyon molekül-l ve von Willebrand Faktörü (vWF) sentezleyip açığa çıkarır.
ECnin yüzeyindeki bu proteinler, trombositleri lokalizeeder ve adezyonununu arttırır ve ayrıca lökositlerin dokulara doğru göç etmesini de sağlar. Açığa çıkan subendotelyal matriks vWF’yi bağlar ve kolajen, trombospondin ve fibronektin gibi diğer prokoagülan adezif moIekülleri de içerir. Bu moleküller hem trombositleri tutmak için ligandlar olarak, hem de yapışık trombositlerin aktivatörleri olarak görev yaparlar. Özellikle, güçlü bir trombosit agonisti olan kollajen trombositlerin yoğun granülerinin boşalmasına ve glikoprotein IIb/IHa (GPUb/IIla; daha sonra açıklanacaktır) gibi aktif ligandların ortaya çıkmasını sağlar. EC hasarıyla ortaya çıkan bir başka önemli prokoaglilan aracı ise subendotelyal düz kas hücreleri ve fibroblastlar tarafından salınan doku faktörüdür (TF). Daha sonra da açıklanacağı gibi (bkz. Koagülasyon basamakları) TF,fibrin pıhtısının oluşmasıyla sonuçlanan çözünür koagülasyon sisteminin en önemli başlatıcısıdır. ECnin ve subendotelyal matriksin bu prokoagülan özellikleri, endotel hasarının kapanmasını ve kanamanın durmasını sağlar. Hasarın etrafında bulunan normal ECler ise bu sırada antikoagülan özellikler göstererek bütün damarda tromboz oluşmasını engeller. Antikoagülan fonksiyonlar daha önce de belirtildiği gibi prostasiklin ve nitrik oksit ile oluşturulabilir ya da damar harabiyeti ile veya pıhtılaşma basamaklarının kendisi tarafından başlatılabilir. Endotelyal hasar veya pıhtılaşma bölgesinde oluşturulan trombin, normal EClere difüze olur ve burada yüzey trombomodüline bağlanır. Trombin trombomodüline bağlandığında, primer çözünür prokogülan bir faktör olmaktan daha çok doğal antikoagulan sistemin başlatıcısı olarak hareket eder. Trombin-trombomodülin kompleksi protein Cyi aktive olmuş formu olan APCye çevirir, koenzimi Protein S, Ya ve YIIIa faktörlerini daha fazla trombin oluşmasını önlemek için inaktive eder. Bununla beraber EC doku-tipi plasminojen aktivatörünün (t-PA) salgılanmasını sağlar, fibrinolizin primer başlatıcısıdır ve plasminojeni, fibI’in pıhtılarını parçalayanaktif enzim plazminine dönüştürür. Doku plazminojen aktivatörü (t-PA) etkisini tam gösterebilmek için pıhtıya direk olarak bağlanabilmelidir. Böylece pıhtının şekillendiği bölgede fibrinolitik cevabın sınırlanmasına yardım eder.
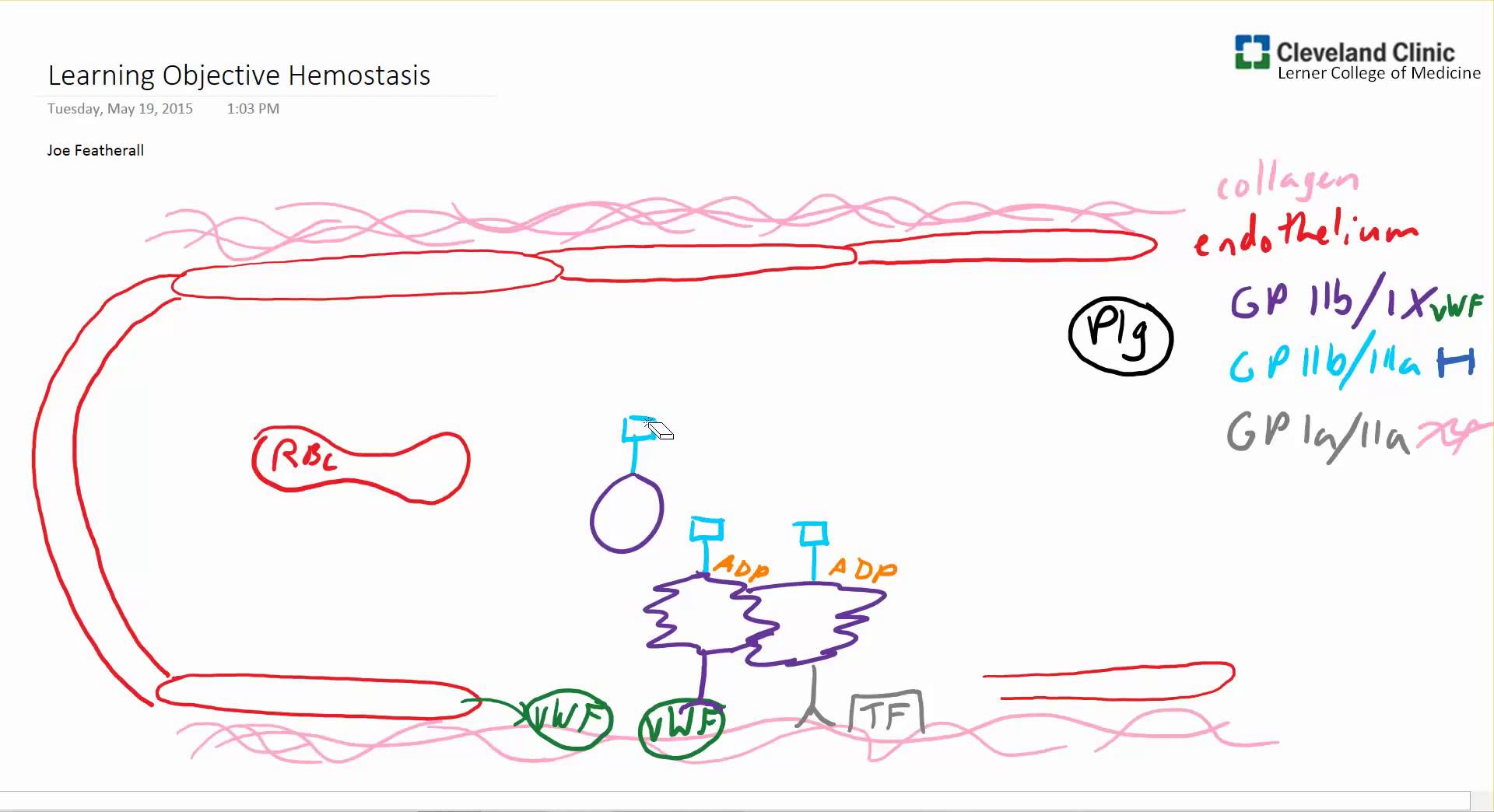
ECler; ayrıca, trombositten salınan adenosindifosfatı (ADP) parçalayan adenosindifosfataz (ADPaz) salgılar ve bu da ilave trombosit aktivasyonunu baskılar böylece de pıhtının büyümesi engellenir. Ayrıca ECIer, hem TF yıla prokoagülan kompleksini, hem de TF’nin neden olduğu Xaz kompleksini (daha sonra bahsedilecektir) çok güçlü şekilde baskılayan TF yolu inhibitörünü (TFPI) salgılarlar. TFPI, Faktör Xa ve TF ile kompleks yaparak aktivitelerini baskılar, trombin oluşumunu azaltacak şekilde etkiler. Damarın prokoagülan ve antikoagülan özelliklerinin dengesi plateletleri ve çözünür koagülasyon faktörlerini lokalize eder ve aktivasyonlarını düzenler.Trombosit, hemostaz için hücresel- temelli bir platform gibi görev yapar. Trombosit membran reseptörleri primer hemostaza aracılık eder ve trombositlerin hasar yerindeki endotele ve subendoteldeki yapılara doğrudan bağlanmalarına izin verir. Trombosit adezyonu, trombosit aktivasyonu oluşturmak için yüzey reseptörleri aracılığıyla transmembran sinyallemeye neden olur. Trombosit adezyonu ayrıca membran yüzeyine reseptörlerin translokasyonu, reseptör yapısal değişikliği, granül içeriğin salımını ve membran fosfolipidlerinin açığa çıkması ile daha ileri prokoagülan fonksiyonlara neden olur. Trombositin prokoagülan yüzeyi, daha sonra koagülasyon basamaklarının kurulması ve trombin oluşması için bir platform olarak görev yapar. Bu platform prokoagülan cevabı artırmak için platelet üzerinde ve pıhtılaşma basamaklarında geri bildirimler yapar, uzun etkili sekonder hemostazı oluşturmak için fibrin üretir. Plateletler, son olarak pıhtı ortarmnda faktör XIII ile pıhtımn pekiştirilmesİne ve platelet faktör 4 ile pıhtının fibrinolizden korunmasına yardım ederler. Dolaşımdaki plateletler 2-4 om çapında, 6-11 L. hacminde, çekirdeksiz hücrelerdir. Plateletler, aşağı yukarı 4 günlük bir olgunlaşma dönemi sonunda megakaryosit stoplazmasından türetilirler ve her bir megakaryosit yaşamı boyunca ortalama 1000 trombosit oluşmasına neden olur.
Trombositler dolaşıma karışır, 7 ila 10 gün yaşarlar, daha sonra ömürlerinin dolması ve vasküler yapısal bütünlüğün sağlanması amacıyla dolaşımdan uzaklaştırılırlar. Vasküler yapıların bozulmadığı (ameliyat sonucu bozulabilmektedir) ve ek stresörlerin trombosit tüketimine neden olmadığı (örn., Sepsis gibi) durumlarda vasküler yapısal bütünlüğün sağlanması için her gün ortalama 7100 trombosit/mcL gereklidir. Normal trombosit sayısı 150,000 ila 450,000/mcL arasındadır. Trombosit sayısı normal sınırlar içinde ve trombosit fonksiyonu normalolduğunda, trombosit fonksiyonunu ölçen kanarna süresi 8 dakikadan daha azdır. Trombosit sayısı 100.000/mcL.'nin altına düşmediği sürece, tek başına trombositopeni kanama süresini uzatmaz. Ancak trombosit sayısı 100.000/mcL. altına düştüğü zaman, trombositopeninin veya anormal platelet fonksiyonunun ya da anormal damar adezyonunun neden olduğu kanarnalarda kanama süresi farklılık göstermez. Trombosit sayısı 100.000/mcL. altında iken benzer şekilde in vitro kanama süresi testi (Trombosit fonksiyon analizi: [PFA]- 100) de trombositopeni veya anormal trombosit fonksiyonu arasındaki farkı belirleyemez. Trombosit-damar çeperi etkileşimi, en iyi şekilde, yüksek akış hızına sahip arteriyel dolaşımda gözlemlenebilir. Farklı hızlarda hareket eden kan paralel düzlemler oluşturur ve damar çeperinin yakınındaki kan, merkeze yakın kandan daha yavaş hareket eder. Bu farklı hızlar ise damar duvarında en büyük shear stresi (akım hızındaki fark) yaratırken, damar merkezine doğru stresin şiddeti azalır. Akım hızı farkı damar çapına ters orantılı olarak değişir, büyük arterlerde 500/sn ve en küçük arteriyollerde 5000/sn şeklinde ölçülmüştür. Aterosiklerotik plakların yüzeyindeki akım hızı farkı ılırnIı stenozda (%50) 3000’den 10,000/sn’ye kadar değişir, hatta klinik olarak ciddi stenozlarda daha da yüksektir. Yüksek hızdaki arteryel kan akışı; (1) prokoagülan reaksiyonların oluşması için gerekli zamanı kısıtlayarak ve (2) hücreler ile proteinlere hasar verip onların damar çeperlerine sıkıca tutunmalarını engelleyerek pıhtılaşmaya direnç gösterir. Bununla birlikte; bir kez damar çeperleri hasarlandığında ve kanama başladığında; trombositler hızla ve kararlılıkla endotelyal bütünlüğün kaybına tepki verir, aynı zamanda da akımla sürüklenip uzaklaşmalarına da karşı koyarlar.
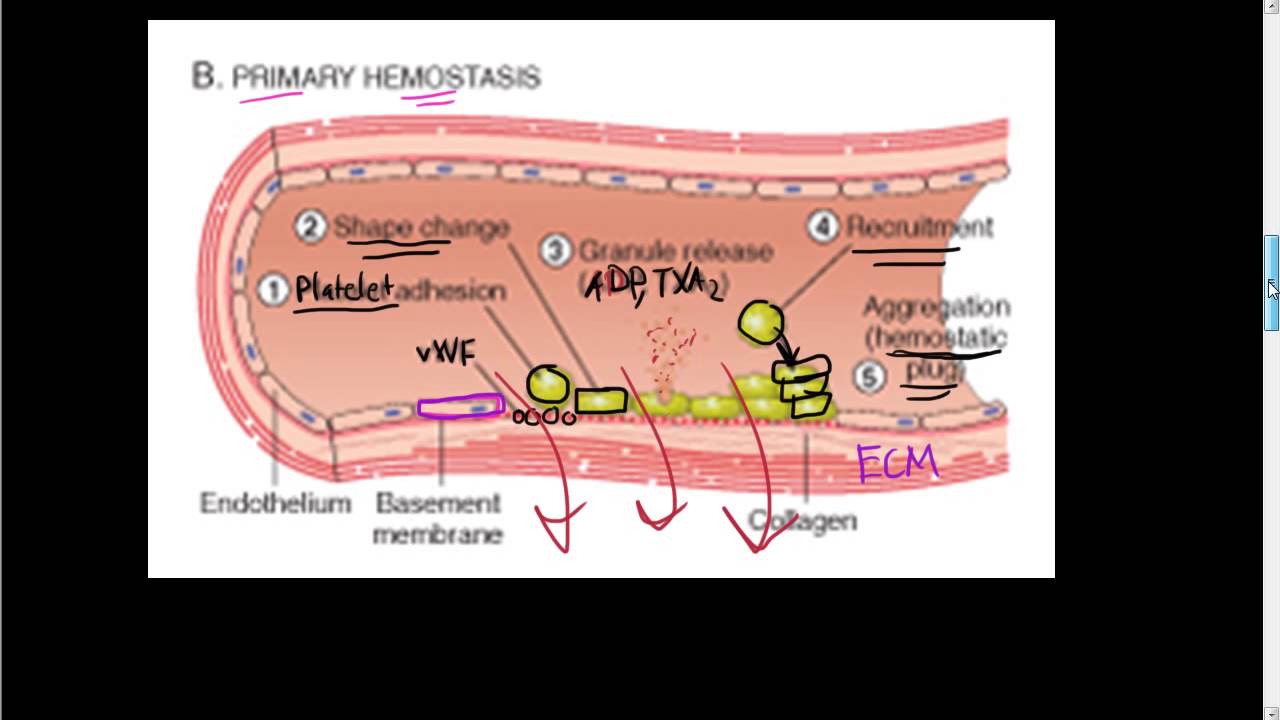
Trombositleri arteriyal dolaşımda çeper adezyonuna hazırlayan güçlerden biri de daha büyük hücreleri (eritrositler ve lökositler) akış hızı farkının en düşük olduğu damar merkezine doğru hareket ettirme eğilimi olan arterial dolaşımdaki kan akışının radyal dağılımıdır; bu yol daha küçük trombositleri damar çeperine doğru iter ve hemostatik mücadeleye tepki verebilecek konuma getirir. Bu boyut-bağımlı akış; ayrıca; kırmızı kan hücre transfüzyonlarının ciddi üremi hastalarında görülen şiddetli anemiyi düzelterek kanamayı yavaşlatma veya durdurma yetisinin paradoksunu da açıklar. Bu etki; bir yandan da arteryal hemostazda trombositierin önemini de ortaya koyar; trombositlerin sayısı veya fonksiyonundaki düşüşler çok ciddi arter kanaması ile ilişkili olabilir. Diğer taraftan; venöz dolaşımda akış hızının daha düşük olması, hücrelerin rastgele hareket etmesini ve koagülasyon reaksiyonlarının daha uzun bir zamanda gerçekleşmesini sağlar, trombosit sayısı ve fonksiyonunun önemi azalır. Bir arteryal kanama yerindeki yüksek hıza sahip kan akışının ayarlanması için trombositler hasar görmüş damara anında yapışmalı ve aktive olmalıdır. Subendoteliyumda bulunan iki molekül; vWF ve kolajen, bu süreçte çok önemlidir. Akış hızı çok fazla olan damarlardaki kanamanın kontrol edilmesi tamamen vWF molekülünün varlığına ve fonksiyonuna bağlıdır. vWF; EClerde ve megakaryositlerde multimerik diziler şeklinde sentetik edilen büyük bir moleküldür ve hem kana salınır hem de Weibel-Palade cisimlerinde depolanır. Çok büyük vWF multimerleri, özeııikle immobilizasyondan sonra açılmamış haliyle, faktör VIII ve trombositleri bağlamada en etkilidirler. Yüksek akış hızlarında, açığa çıkmış subendotelyal kollajene GPlb reseptörü kompleksi ile yapışarak immobilize olan vWfnin en büyük multimerik farınları trombasit yüzeyindeki GPV ve GPIX’ye de bağlanır. Bu bağlanma oldukça hızlıdır fakat bu yüzeyde trombasitleri yavaşlatan düşük afiniteli bağlanma onların sadece subendatelyal vWF’ye hafifçe tutunmalarıyla sağlanır. Plateletler artık dolaşımda rahatça akmayıp subendotel üzerinde toplandığı için yüksek akış hızlarında GPIb- V-IX-vWF etkileşimi ile oluşan transmembran sinyalıerne platelet disk şeklinin kaybına ve bir başka platelet reseptörü olan GPUb/IIIa da yapısal değişikliğe neden olur. Aktive olan GPUb/Illa reseptörü GPlb bağlanma bölgesinden uzak bir yerde ya fibrinojene ya da büyük vWF multimerlerine bağlanır. Bu ikinci tür adezyon, GPlb- V-IX-vWF bağından daha yüksek afiniteli etkileşime sahiptir ve trombositi subendoteliyuma sılaca bağlar. vWF aracılığıyla trombosit bağlanması ve aktivasyonu sürecinin önemli bir düzenleyecisi de; vWFparçalayan proteazdır. Trombospondin tip i motifli disintegrin ve metalloproteinaz olan ADAMTS-13 son derece büyük olan multimerleri, platelet bağlanması için daha küçük parçalara (platelete bağlanma afinitesi azalmış) ayırmak suretiyle vWF aktivitesini düzenler. Parçalayıcı proteaz aktivitesinin kaybı son derece büyük multimerlere kontrolsüz platelet bağlanmasına ve mikrovasküler tromboza (trombotik trombositopenik purpuraya )neden olur.
Daha ılımlı akış hızlannda GPlb-V-IX-vWF adezyonu, subendotelyal kollajenin bir başka yapışkan kısmına, GPlalIla 'ya plateletin bağlanmasıyla desteklenir. Dolayısıyla subendotelyal vWF ve kollajen platelet adezyonunu başlatmada işbirliği yaparlar ve yüksek akış hızlarında subendotelyal vWF basıan roloynar. Kollajen GPlalIIa’ya plateletleri bağlayarak plateletleri bir noktada tutabilmeleri ve ikinci bir bölgede GPVI’e plateletleri bağlayarak platelet aktivasyonunu yapabilmeleri açısından eşsizdir. Önemli platelet adezyon reseptörleri; GPIIbIIIIa, GPfbV/lX, GPVI veya GPlalIla’nın herhangi birinin konjenitaj olarak eksikliği ciddi hemostatik bozukluğa neden olur ve sadece platelet transfüzyonu ile tedavi edilebilir. Aynı şekilde, vWF de azalmalar özellikle daha büyük multimerik formlarda olduğunda kanamaya neden olabilir. Bir platelet tabakası kanarna bölgesine yapışır yapışmaz, vWF, küme oluşturmuş plateletlerin en üstünde yer alan GPIb, V ve lX’a bağlanır, büyüyen platelet tıkacının içine akan kanla gelen plateletler de işleme katılır. Bağlanan plateletler daha sonra “aktivasyon” adı verilen birbirine bağımlı bir dizi süreçten geçerler. Platelet aktivasyonunun beş önemli etkisi vardır; I) platelet-platelet matriksinin stabilizasyonu için gerekli olan ligandıarın lokal olarak salınımı 2) ilave plateletIerin temininde devamlılığın sağlanması 3) yavaş kanamalarda daha küçük arterlerin vazokonstriksiyonu 4) platelet eşliğinde fibrin oluşumunun lokalizasyonu ve hızlandırılması 5) pıhtının fibrinolize karşı korunması. Platelet tıkacının aslı köprü kurucu ligandlar olan fibrinojen ve vWF ile platelet-ligand-platelet matriksinin oluşmasıdır. Fibrinojen ve vWF’ün her ikisi de uyarılmamış plateletin içindeki alfa-granüllerde depolanır ve platelet aktive olduğunda buradan salınır. Her ikisi de iki platelet üzerinde yeralan GPIIb/Illa reseptörüne bağIanabilir, ve bunları birbirine bağlayabilir. Daha önce bahsedildiği gibi platelet GPIIblIIIa reseptörü kalsiyuma bağımlı olarak yapısal değişikliğe uğrar, bu değişimde reseptörün fibrinojen ya da vWF üzerinde bir amino asit hajkası arjininglisin aspartat (RGD) içeren bir bölgeye bağlanmasını sağlar. Her bir fibrinojen molekülünün polar uçlannda iki RGD bölgesi vardır. Daha geniş vWF multimerleri birden fazla RGD bölgesi içerir. Hepside yapısalolarak değişmiş GPUb/Illa’ya bağlanabilme kapasitesindedir ve platelet-ligand-platelet matriksini oluşturur.
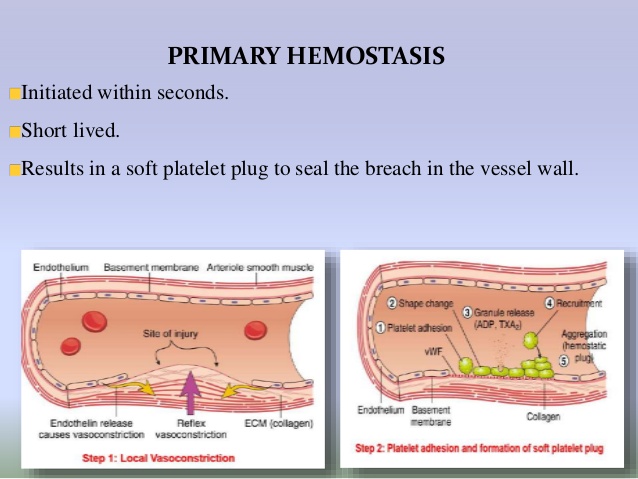
GPUblIna, platelet yüzeyinde en fazla bulunan glikoproteindir, uyanlmamış plateletlerde yaklaşık 50.000 kopyası vardır, sitozolde bulunan GPUb/Illa reseptörleri, plateletin aktivasyonundan sonra yüzeye doğru hareket eder. Kollajen, epinefrin, trombin gibi lokal agonistler plateletlerin platelet tıkacı içine katılmalarını sağlar, plateletler de lokal mikroçevreye agonistleri salgılar. Plateletleri güçlü bir şekilde aktive etmek için kollajen (daha önce bahsedildi) ve trombinin her ikisi de platelette bulunan kendilerine ait özel reseptörleri ile etkileşirler. Her ne kadar epinefrin tek başına güçlü bir platelet agonisti olmasa da platelet üretimindeki a-adrenerjik reseptörün uyaniması onları ADP gibi nisbeten zayıf agonistler tarafından bile sinerjistik olarak aktive olmaya hazırlar. Plateletden doğrudan doğruya salgılanan tromboksan Aı (TxAı) araşidonik asidin siklooksijenaz ile parçalanmasInı takiben sitozolde şekillenir ve daha sonra pıhtı ortamına salınır.
TxAı hem platelet agonisti hem de vazokonstriktördür, hızla inert ürünü olan tromboksan Bı’ye parçalanır. Platelet sikjooksijenaz- i (COX-I) aktivitesi aspirin ile geri dönüşümsüz spesifik bir serin ile dönüşümsüz,kovalent bağ oluşturur. Steroid olmayan antienflamatuar ilaçlar (NSAIDs) serindeki asetilasyondan dolayı kovalent olarak bağlanamaz. Bunun yerine aktif, katalitik trozin bölgesinden geriye dönüşümlü olarak ve yanşarak bağlanırlar. Dolayısıyla NSAlD antiplatelet etkileri, aspirinin tersine ilacın plazma düzeylerine bağlıdır. Olgun plateletler COX-2 aktivitesine sahip değildir, bundan dolayı enflamatuar hastalıklar için oldukça seçici COX-2 inhibitörleri geliştirmenin prensiplerinden biri, platelet COX-l aktivitesini etkilemeksizin platelet işlev bozukluğunun sebep olduğu kanamalardan kaçınmaktir. Ne yazık ki klinik çalışmalarla, bir kısım seçici COX-2 inhibitörlerinin, muhtemelen antitrombojenik bileşik prostosiklinin oluşumunu bloke ederek miyokard enfaktüsü ve inmede dahilolmak üzere hipertansiyon ve vasküler olaylar olasılığını artırdığı gösterilmiştir. Diğer platelet agonistleri, yoğun ve alfa granüllerin plateletin kanaliküler membranı ile birleşmesi sonucunda hücre dışı sıvıya salınırlar ve böylece granül içerikleri dışarı atılmış olur. Yoğun granül içeriğinden olan serotonin, TxA2’ye benzer, her ikisi de platelet agonistidir ve vazokonstriktördür. Bir başka yoğun granül bileşiği olan ADP tamamen vazoaktif özellikleri olmayan bir platelet agonistidir. TxA2 ve serotonin aracılıklı vazokonstriksiyonun önemi tamamen açık değildir. Bununla birlikte damar çapını azaltarak oluşturulan vazokonstriksiyon kanın akış hızını artırabilir ve böylece plateletlerin hasarlı alana sevk edilmesini kolaylaştırabilir. Yoğun granül salınımının hemostazın sürdürülmesindeki önemi konjenital yoğun granül yetmezliğinde görülen şiddetli kanama ile vurgulanmıştır (örn., Hermansky-Pudlak sendromu). Bundan dolayı platelet aktivasyonu, platelet adezyonunu şiddetlendirir ve koagülasyon basamakları ile etkileşerek fibrin üreten prokoagülan aktivite için platelet yüzeyinin uygun hale getirilmesini sağlar.
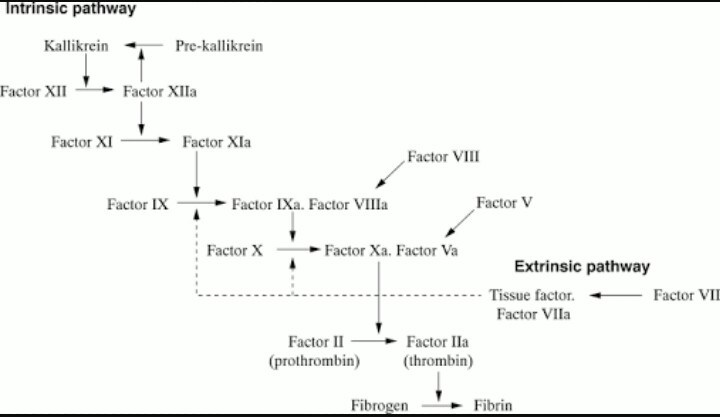
Koagülasyon basamakları, dolaşımdaki antikoagülan proteinlerle kontrol altında tutulan sürekli faktör aktivasyonu ve enzim kompleksIerinin koordineli bir şekilde toplanmasıyla karakterizedir. Bu enzim kompleksIeri fosfolipid membran üzerinde toplanmış serin proteazları, kofaktörleri ve zimojen substratlan içerir. Normal şartlarda bu kompleksIerin oluşumu ve ortaya çıkan trombin üretimi nispeten yavaş olur. Dolaşımdaki antikoagülanlar ile kompleksierin inaktivasyonu bunlann prokoagülan aktivitelerini dengeler ve pıhtı oluşumunu engeller. Ancak aktif faktör oluşumunu tetikleyen bir prokoagülan uyarı ortaya çıktığında, bu enzim kompleksIerinin oluşumu hızla artar ve yoğun trombin üretilir ve arkasından da fibrin oluşur. Koagülan faktörlerin çoğunun başlıca sentezlenme yeri karaciğerdir. Şiddetli karaciğer hastalıklannda faktör VIII dışında tüm koagülasyon faktörlerinin düzeyleri düşer. Bu da faktör VIII’in sadece karaciğer tarafından deği] aynı zamanda ECler ve retiküloendotelial sistemin hücreleri tarafından da üretildiğini düşündürür. Ayrıca faktörlerin bir alt kümesi vitamin K bağımlıdır, bunlar protrombin (faktör II), faktör VII, IX, ve X’dur. Doğalolarak oluşan antikoagülanlar yani protein C ve S:de vitamin K’ye bağımlı olarak sentezlenir. Bu proteinlerin amino-terminal bölgelerinin posttranslasyonal değişimi (bir vitamin K -bağımlı karboksilaz aracılığı ile) 10-12 kadar y-karboksiglutamat kalıntısı eklenir. Bu kalıntılar, kalsiyum bağlanması, proteinlerin üç boyutlu yapılarının saptanması, membran yüzeyine uygun bağlanmalann yönlendirilmesinde önemli roloynar. Warfarin vitamin K’nın karaciğere alımını bloklar ve bu karboksilazın fonksiyonunu baskılar. Laboratuar testleri açısından koagülasyon basamakları suni olarak ekstersek (protrombin zamanı [PT] ) ve intrensek (kısmi tromboplastin zamanı [PTT] ) olmak üzere ikiye ayrılır, her ikisi de ortak yolda buluşur. Bu ortak yol trombin ve fibrin üretimidir . Bununla beraber, anlaşılması gereken önemli bir nokta da fizyolojik koagülasyonun, kompleks faktör etkileşimlerinin tek bir yol olmasıdır.
Koagülasyonun fizyolojik başlatıcısı TF’dir. TF, ancak EC hasarı ile kana maruz kalan subendotelial fibroblastlar ve düz kas hücrelerinde oluşturulur. TF ayrıca endotoksin gibi aktive edici veya enflamatuar uyarılara maruz kaldıktan sonra periferal kanın monositleri ve vasküler ECler üzerinde ortaya çıkar. Laboratuarda ekstrinsek yol dolaşımdaki faktör VIIa ile dışarıdan eklenen TF-tromboplastin etkileşmesinin ölçülmesiyle değerlendirilir ve PT (protrombin zamanı) ölçülür. Protrombin zamanı VII, V, X ve protrombin (faktör II) eksikliklerine karşı oldukça duyarlıdır ve tüm bu eksiklikler önemli kanama komplikasyonları ile ilişkili olabilir. Faktör VII en kısa yarı ömre sahip olmak üzere faktör II, VII, ve X, vitamin K bağımlısı olduğu için, PT varfarinin (coumadin) tedavi etkinliğinin en hassas ölçüsüdür. PT, faktör XII, XI, IX veya VIII eksikliğinden hiç etkilenmez. Varfarin ile protrombin zamanının uzama derecesi kısmen testte kullanılan tromboplastinin gücüne bağlıdır. Bu güç ise, tromboplastinin her bir test aletinde gösterdiği kendine özgü aktivitesi ve imalatçısına göre değişir.
Bu nedenle PTde varfarin tarafından meydana getirilen değişiklikleri laboratuarlar arasında standardize edip global olarak antikoagülan önerilerinin uygulanabilmesini sağlamak amacıyla uluslararası normalize oran (international normalized ratio-INR) oluşturulmuştur. INR her tromboplastinin uluslararası duyarlılık indeksine (ISI) dayanır, TF olarak standardize edilmiştir ve her hasta için şu şekilde hesaplanır: (hastanın PTsilortalama kontrol PT). Varfarine zıt olarak, terapötik düzeylerde PT unfraksiyone heparine göreceli olarak duyarsızdır. Hematoloji laboratuarında koagülasyon yolunun in vitro temas aktivasyonu PTT olarak ölçülür; test kaolin gibi negatif yüklü bir bileşikle plazmanın uyanlması ile başlar. PTT çalışması temas faktörlerinin (prekallikrein [PK], yüksek moleküler ağırlıklı kininojen [HMWK], ve faktör XII), ve koagülasyon faktörleri XI, IX, VIII, V, X ve plazminojen eksikliklerine duyarlıdır. PK, HMWK ve faktör XII eksiklikleri klinik olarak kanamaya yol açmaz, bu da bu özel in vitro koagülasyon başlatıcılarının fizyolojik hemostazia ilgisiz olduklan anlamına gelir. Buna zıt olarak, faktör XI, ve özellikle de faktör IX ve VIII eksiklikleri, genellikle önemli kanamaya neden olur. PTT unfraksiyone heparine yüksek derecede duyarlıdır ve terapötik heparin düzeylerinin hızlı izleme çalışması olarak kullanılır. INR’nin varfarin (Coumadin) için olduğu gibi olmayıp, unfraksiyone heparin için terapötik PTT düzeylerinin yelpazesi daha geniştir ve iyi standardize edilmemiştir. Terapötik unfraksiyone heparin düzeyleri (anti-Xa aktivitesinin çalışılması ile ölçülür) genellikle hastanın başlangıç PTT’sinin (heparin başlanmadan önceki) ya da kontrol popülasyonunun ortalama PTT’sinin 1.8 ile 2.5 katı arasına tekabül eder. Daha önce belirtildiği gibi, intrinsik ve ekstrinsik koagülasyon yolu şeması sadece in vitro test amacıyla makul bir paradigmayı yansıtır. Fizyolojik olarak pıhtılaşma basamakIannın başlamasına doku hasan ve dolaşan aktive olmuş faktörlerin düşük düzeyleri yol açar, ve koagülasyon olayları ayrıca enflamasyon la iki yönlü ilişkilidir; aktive olmuş makrofajlar işlevsel TF’yi açığa vururlar, ve faktör Xa enflamatuar yanıtları indükler. in vivo olarak koagülasyon basamakları fosfolipid membran yüzeylerinde aktif olarak işlev yapan enzim kompleksIeri tarafından üretilir.
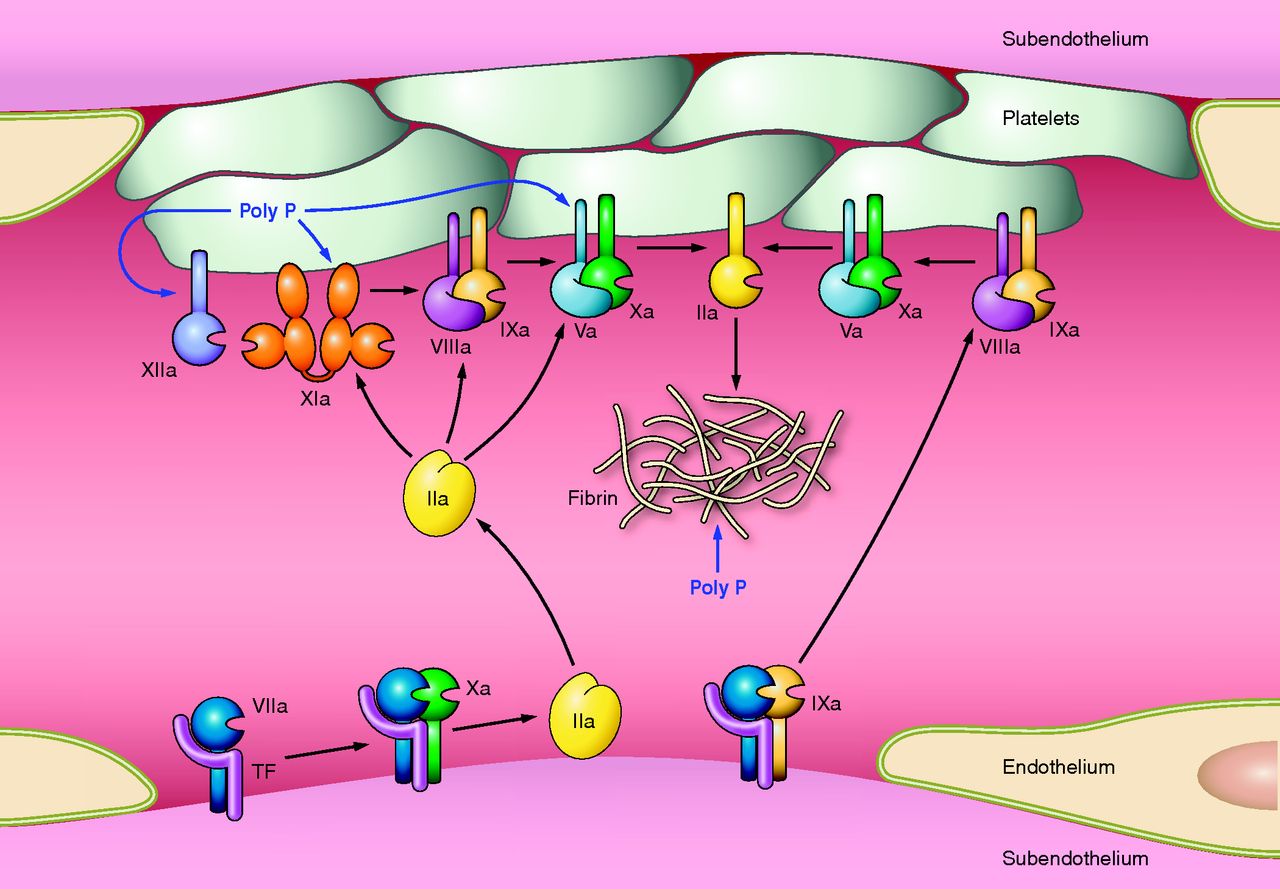
Uygun fosfolipid yüzeyinin baskın olarak aktive trombositler ve ECler tarafından temin edildiği görülür. Dolaşan faktör YIIa’nın düşük düzeylerini trombin ve faktör Xa-fosfolipid kompleksi ve daha düşük oranda da faktör YIIa-TF’nin kendisi oluşturur. EC hasarından sonra ortaya çıkan TF dolaşan faktör Yıla’ya bağlanır ve faktör Ylla-TF kompleksi aktive trombosit membranında ardışık olarak zimojen substratları olan faktör IX ve X’a bağlanır ve TF’ye-bağımll tenaz kompleksini oluşturur. Tenaz ve protrombinaz reaksiyonu (daha sonra açıklanacak) için gerekli negatif fosfolipid membranı oluşturmaya ek olarak, aktive trombositler faktör Xa, IXa, ve Ya için özgül reseptörler temin ederler. Aktive trombositin alfa granülünden aynı zamanda faktör Y salınır, buna rağmen kanıtlar salınan faktör V’in çoğunun plazma havuzundan türediğini göstermektedir. Bu koagülasyon faktörlerinin negatif yüklü, trombositin açığa çıkardığı fosfatidilserinle olan uzaysal yerleşimlerinin membranla ilişkisi, prokoagülan enzimatik reaksiyonları hızIandırır ve eş zamanlı olarak dolaşan inhibitörlerden aktive faktörleri korur; bu işlem hızlanmış trombosit üretiminin doruğa ulaşmasını sağlar. TF faktör YTla trombosit resptörlerine bağlanmış olan faktör X’u Xa’ya, faktör IX’u IXa’ya dönüştürür. Trombositin faktör Xa reseptörünün tmmbosite bağlı faktörYa ile sıkı ilişkisi vardır; bu faktörler serbest kalsiyum ve trombosit membran fosfatidilseriniyle birlikte protrombine (faktör 2) bağlanıp protrombinaz kompleksi oluştururlar, böylece her ne kadar küçük miktarda da olsa trombin oluştururlar. Başlangıçta oluşan bu trombin önemli miktarda fibrin oluşturmayabilir, fakat bunun yerine trombosit reseptöriine bağlanan membrana bağlı faktör Xa, faktör V ve Va’ya bağlanan faktör XII ve Xla’yı aktive edebilirler. Bu noktaya kadar, faktör Xa ve trombin üretiminin oluşumu göreceli olarak yavaştır.
Bununla beraber, başlangıçtaki bu trombin, trombosit yüzeyinde fark edilir miktarda faktör Xla ve Va ve kofaktörleri oluştuğunda, özellikle dolaşan TFPI TF’nin işlevini süratlice bloke ettiğinde, faktör X’un kinetik olarak müsait yolda aktivasyonu baskın olur. Trombinin geri besleme aktivasyonu yoğun olarak pıhtılaşma basamaklarını artırır, böylece faktör Xa ve trombin üretiminin oranı katsayısalolarak artar. Daha önceden belirtildiği gibi, bu üretim başlangıçta TF’nin indüklediği faktör lXa ve X’un oluşturduğu Xaz kompleksinin, ve daha sonra faktör IX’un uzatmalı faktör Xla’ya dayalı aktivasyonu ile tenaz oluşumu aracılığıyla olur. Faktör Xa kofaktörü olan faktör yına ve zimojen substratı olan faktör X’a bağlanır ve membran yüzeyinde faktör Xa oluşumunu daha fazla artırır. Takip eden faktör Xa üretiminin patlaması, protrombinaz kompleksinin oluşumunu artırır ve daha da yüksek oranlarda trombin üretimi ile sonuçlanır. Trombin üretimindeki bu hızlı artışın sonucunda fibrinojen fibrin monomerlerine ayrışır ve bütün bu monomerler hızla birleşerek platelet tıkacı ile bütünleşmiş olan fibrin matriksini oluşturur. Trombinin hem plazmada bulunan hem de plateletten salman XIII üzerine etkisi ile oluşturulan bir transamilaz olan faktör XIlIa, çözünür fibrin pıhtısını çözünmeyen fibrm polimerine çevirir ve aynı zamanda pıhtının plazmin aracılıklı parçalanmasını önlemek için fibrini, plazmine bağlar. Son olarak platelet tıkacı, pıhtının kasılmasına maruz kalır, bu kasılmada platelet-fibrin matriksini plazmin tarafından oluşacak lizize karşı korur. Büyük ölçüde platelet aktivasyonu ile ilgili olan bu antilitik mekanizmalar plateletlerce zengin pıhtıların trombolizise karşı olan göreceli direncini açıklayabilir. Platelet ve EC membranIarı üzerinde faktör X’u parçalayan enzim (tenaz) ve protrombinaz kompleksieri oluştuğu anda koagülasyonun doğal inhibitörleri de pıhtılaşmayı azaıtacak şekilde etkilemek için aktif hale gelirler. Hasar görmemiş dolaşımda ve yeni oluşmuş pıhtının çevresinde bulunan bulunan hasarsız bir EC bariyerinde bulunan mevcut endojen mekanizmalar pıhtılaşmayı hasar bölgesinde sınırlar ve etrafında akan kanın sıvı kalmasını sağlar. Arteryel dolaşırnda büyüyen pıhtının neden olduğu damar oklüzyonuna koagülasyon faktörlerini seyrelten ve dağıtan yüksek hızlı kan akışı ile karşı konulur. Antiplatelet faktörlerde sağlıklı EC tabakasının özünde olan antikoagulan aktivitenin bir parçası olup, platelet tıkacının hasar bölgesi dışına taşmasını sınırlamaktadır. Bu faktörler şunları içine olmaktadır; i) benzer yüklü plateletleri uzaklaştıran yüzeyin net negatif yükü 2) platelet kümeleşmesini baskılayan nitrik oksit ve prostosiklin salınımı ve 3) plateletten salınmış ADP’yi etkisiz hale getiren ve böylece ilave plateletlerin görevlendu·ilmesini sınırlayan bir ADpaz’ın oluşup yüzeyde açığa çıkması. Platelet tıkacı ve bununla ilişkili fibrin birikimi kanamayı durdurur durdurmaz ve açığa çıkmış endotel örtülür örtülmez koagulasyon basamaklarındaki dizginleme önemli hale gelir. Pıhtının sınırlanması çeşitli mekanizmalarla olur; 1) TFPI tarafından tenaz ve TF-faktör VIla’nın nötralizasyonu, 2) antitrombin ILI (AT ın)tarafından trombin ve faktör IXa, Xa ve Xla’nın nötralizasyonu, 3) APC ve kofaktörü protein S tarafından trombinin aktive ettiği kofaktör Va ve VlIIa’nın parçalanması ve 4) t-PA ve urokinaz tarafından katı fibrin pıhtısının çözünmesi. AT ın ve TFPI dolaşırnda bulunan proteaz inhibitörlerdir. AT III, trombin ve faktör Xa, IXa ve Xl’nın aktivitesini bu proteinler ile kompleks oluşturmak suretiyle baskılar. AT m’un antitrombin ve anti-Xa aktivitesi parçalanmamış heparin ile yaklaşık 2000 kat arttırılır. ECler bir endojen glikozaminoglikan olan heparin sülfatı sentezler. Heparin sülfat ise ekstraseııüler matriks ile birleşu· ve daha sonra lokal olarak gelişmiş trombinin nötralleşmesini artırmak için kan AT ILI ile kompleks oluşturur. Heparin sülfatlar hasarsız komşu endotel ile ilişkili olan hücre dışı matrikse bağlı oldukları için AT IlI-heparin etkileşimleri pıhtının hasarlı bölge dışına çıkacak şekilde uzamasını engellemeye yardımcı olur. ECler dolaşımdaki faktör Xa aktivitesini de baskılayan TFPI’yl salgılarlar, ancak ilk görevi TF’nin neden olduğu Xaz fonksiyonunu azaltarak etkilemektir. Bu görev TFPI-faktör Xa kompleksinin TF-faktör Yıla kompleksine bağlanması ve bu dörtlü kompleks oluşumu ile TFfaktör VIIa aktivitesinin etkisiz hale getirilmesi ve böylece TF ile başlatılan Xaz yolunun kapatılması ile başarılır.Trombomodulin, EC-yüzeyi ile ilişkili başka bir proteindir.
Antitrombin nötralleşmesinden kurtulan trombin yakındaki hasarsız komşu EC’lerin membranı üzerinde yer alan trombomoduline bağlanır ve bu enzim kompleksi protein C’yi etkin hale getirir. Aktif protein C (APC) plateletle ilişkili olmayan faktörler Va ve Vllla’yı parçalar ve dolayısı ile ilgili protrombinaz ve Xaz kompleksIerini etkisiz hale getirip trombin oluşumunu azaıtırlar. Vitamin K’ya bağımlı faktör olan protein S, APC için bir kofaktör rolü oynayıp, Va faktörü ve VUTa faktörünün uzaklaştırılması için APC’nin biyolojik aktivitesini sırasıyla 20 ve 5 kat arttırır. Protein S, C4b-bağlanma proteini ile kompleks oluşturduğu zaman değil sadece dolaşımda serbest durumda olduğu zaman fonksiyon gösterir. Akut hastalıklarda C4b bağlanma proteini akut faz reaktanı olarak artırabilir. Bundan dolayı protein S’in artmış bağlanımı akut hastalıklarda görülür ve bu süreç serbest protein S düzeyini azaltabilir ve doğal antikoagülan aktivitesinde göreceli bir düşüşe yol açabilir ve prokoagülan durum etkin olur. Endotel ilişkili Fibrinoliz Damar içi fibrinolitik aktivite t-PA ve ürokinaz tip PA (u-PA) gibi plazminojen aktivatörler ve plazminojen aktivatör inhibitör -1 (PAl-I) ve a2-antiplazmin gibi inhibitörler arasındaki bir dengeden kaynaklanır. Fibrinolizin düzenlenmesi endotel yüzeyinde olur. Vasküler EC’ler t-PA ve PAI-l ‘i sentezleyip salgılarlar. Özellikle fibrin pıhtısı varlığında plazıninojenin plazmine aktivasyonu hücre yüzeyi ile ilişkili t-PA tarafından ve daha düşük seviyede olmak üzere nisbeten dolaşıında az miktarda bulunan u-PA ile şiddetlendirilir. Plazmin, fibrin ve matriks bileşenlerinin hücreye yakın bölgelerde parçalanmalarını kolaylaştırır. PAl-I, t-PA aktivitesininbaşlıca in vivo blokeridir. t-PA miktarına bağlı olarak dolaşıma salınan PAI-l büyük konsantrasyonlara ulaşır. Ancak sağlıklı kişilerde PAl-I plazma seviyesinde anlaınlı değişiklik olmaz. Plazmadaki PAl-l seviyesinin değişmesi aynı zamanda PAT- ı geninin polimorfizmi ile de ilişkilidir. 4-G promotor bölge polimorfizmi daha yüksek PAI-I düzeyleri ile ve belkide daha yüksek bir tromboemboli riski ile ilişkilidir. Plazminin prokoagülan etki oluşturan inhibisyonu a2-antiplazmin ve muhtemelen a2-makroglobülin aracılığı ile olur. Fibrinoliz için EC’ler yanı sıra makrofajlar da kritik role sahiptir. Makrofajlar plazmin gerektirmeyen bir mekanizmayla lizozomal proteoliz ile fibrin pıhtısını parçalar. Makrofaj fibrine (ogen) yüzey integrin reseptörü CDllb/CDI8 aracılığı ile bağlanır. Bu bağlanmayı kompleksi n lizozom içine alınması (internalizasyonu) takip eder, lizozom içinde de fibrin parçalanır. Dokunun tamiri ve yenilenmesi eninde sonunda fibrin bazlı pıhtının çözünmesini gerektirir. t-PA ve ürokinaz dolaşımdaki zimojen plazminojen üzerine etki ederek aktif fibrinolitik enzim olan plazmine dönüşmesini sağlar. Ayrıca intrinsek yol aktivatörleri olan kallikrein, Xlla ve Xla’ da plazminojenin plazmine dönüştürülmesine yardımcı olurlar. Hücre yüzey reseptörlerine bağlı olan plazminojen oluşan plazmini dolaşımdaki urantiplazmin tarafından etkisiz hale gelmesinden koruduğu gibi t-PA ve fibrin pıhtısına yakın bir konuma getirerek kendi aktivasyonunu da hızlandırır. Plazmin fibrin matriksini çözer ve çözünür fibrin peptitleri ve D-dimer oluşur. Plazmin aynı zamanda hasarlı dokuyu parçalayan metalloproteinazları etkin hale getirir. Fibroblastlar ve lökositler yara içine göç ederler, lökositlere E ve P selektin bağlanma aracılık eder. Bu hücreler lökositlerden ve aktif hale gelmiş plateletlerden salınan büyüme faktörleri ile uyumlu olarak damarın onarımı ve dokunun yenilenmesi için çalışırlar.
Kanamanın değerlendirilmesi; dikkatli bir hikaye, fizik muayene ve laboratuar tetkjklerini gerektirir. Hasta hikayesi; kanama şekli (epistaksis, menoraji, hematom oluşumu vb.), kanamanın hangi durumlarda oluştuğu (travma, cerrahi, dental girişimler vb.) ve kanamayı durdurmak için herhangi bir kan ürününe (ve ne tip bir kan ürününe) ihtiyaç duyulup duyulmadığı hakkında bilgileri içermelidir. Hebm; kanama ile ilişkili olabilecek aspirin gibi bir ilaç kullanımını yada kanamaya yol açabilecek bir enfeksiyon yada karaciğer hastalığını da sorgulamalıdır. Son olarak, ailede kanama öyküsünün araştırılması da önemlidir; hemofili düşünülen bir erkek çocukta , dayılarınca gibi ikinci kuşak akrabaları ve diğer jenerasyonları da incelemelidir. Fizik muayene, kanamanın kökeni hakkında bazı ipuçları verebilir ve peteşial hemoraji gibi küçük damar kanamaları ile hematom ve purpura gibi büyük damar kanamaları arasında ayrım yapılmasına yardımcı olabilir. Deride, mukoz membranıarda ya da gastrointestinal sistemde yer alan küçük damar kanamaları; daha sıklıkla trombositopeni, trombosit defekleri, vasküler anormal durumlar ve von Willebrand hastalığı (vWD) sonucu ortaya çıkma eğilimindedir. Kadınlarda, menoraji tek semptom olarak karşımıza çıkabilir. Solid organlardaki, eklemlerdeki yada kaslardaki büyük damar kanamaları, daha çok hemofili A yada B gibi faktör eksiklikleri ile ilişkilidir. Kanama şikayeti olan hastaların ilk değerlendirmelerinde, laboratuar tarama testleri sıklıkla yararlıdır. Bu testlerde bakılması gerekenler şu şekilde sıralanabilir: (I) kan hücre sayıları (özellikle trombosit sayısı) ve periferik kan smearlerinin incelenmesi; (2) protrombin zamanı (PT), K-vitamini bağımlı koagülasyon faktör eksikliklerine oldukça duyarlıdır; (3) parsiyel tromboplastin zamanı (PTT), faktör VIII, IX ve XI deb eksiklikleri saptamasının yanı sıra bağlantı aktivatörleri, prekallikin, yüksek moleküler ağırlıklı kininogen ve faktör XII eksikliklerine de duyarlıdır. Faktör X, V ve LI (protrombin) düzeylerindeki anormaliteler, hem PT hem de PTT düzeylerinde uzama ile sonuçlanır. Şayet PT ya da PTT süreleri uzamışsa, hastanın plazması normal plazma ile karıştırılır (karışım çalışması) ve pıhtılaşma zamanı tekrarlanır. Bu çalışma, faktör eksikliği (PT ya da PTT normal sınırlarına iner) ile dolaşımdaki inhibitör (pıhtılaşma zamanı halen uzundur.) ayrımını yapmaya olanak sağlar. Kanaması olan hastalarda uygulayabileceğimiz testlerden biri de trombin zamanıdır; bu test direkt olarak eksojen trombin ile fibrinojenden fibrin dönüşümünü ölçer ve hem fibrinojen düzeyini hem de onun fonksiyonel yeterliliğini belirler.
Trombosit fonksiyonları, klasik olarak hastanın derisinde yapılan insizyon sonrası kanamanın durma zamanının invaziv ölçümüne dayanan, in vivo kanama zamanı ile değerlendirilir. Kanama zamanı, trombositopeni (trombosit sayısl<100,000 mcL) ve nitel trombosit defektlerinde uzama gösterir. Kanama zamanı testinin teknik güvenilirliğinin düşük olması ve infantlar ile neonatallerdeki uygulama zorlukları, testin kullanımını sJnırlamaktadır. Günümüzde trombosit fonksiyonlarını in vitro değerlendirmeye yönelik bazı ticari aletler mevcuttur. İn vifro kanama zamanını gösteren aletlerden biri olan Platelet Function Analyzer- 100 (PFA- 100) de; sitrat antikoagule tam kan küçük bir açıklıktan epinefrin, adenosindifosfat (ADP) ve kollajen gibi trombosit aktivatörlerinin bulunduğu bir kartuşa geçer. Trombositler aktive olup yapıştıkça açıklık giderek kapanır ve trombosit tıkacı tarafından tam okluzyonun gerçekleşme zamanı ölçülür. Aspirin alımı veya vWD gibi plazmada azalmış trombosit bağlanması durumlarında görülen nitel trombosit defektIerinde, kapanma zamanı uzar. Her ne kadar trombositopeni kapanma zamanını, in vivo kanama zamanına benzer şekilde etkilese de; in vitro kanama zamanı testleri, manuel tekniklere bağlı hataların ve invaziv prosedürlerin bulunmaması yönünden daha çok tercih edilmektedir. Uzamış PTT’nin değerlendirilmesinde kullanılan diğer bir laboratuar yöntemi, özellikle ayaktan takip edilen hastalarda, polybrene eklenmesi sonrası yapılan PTT ölçümleridir; intravenöz alınan heparinle kontamine edilmiş örneğe eklenen polybrene heparin etkisini nötralize ederek uzanuş PTT zamanını düzeltir. Polybrene PTT, tedavi amacıyla fraksiyone olmayan heparin (UFH) alan hastalar için kullanışlı değildir. Karışım çalışmaları ile düzelmeyen uzanuş PTT, aynı zamanda lupus antikoagulanlı (sıklıkla trombosis içeriğinde) hastalarda da görülür. Bu durumlarda lupus antikoagulant teşhisi; antifosfolipid antikorlarla bağlanacak aşırı fosfolipid eklenmesi sonrası PTT’nin düzelmesi ile olduğu kadar, lupus antikoagulant spesifik diğer testlerle de doğrulanır. Kanamanın olası nedenlerini tanımlamaya yönelik hızlı bir yaklaşımda şu major hastalık kategorileri dikkate alınmalıdu: (1) trombositopeni yada anormal trombosit fonksiyonları; (2) K-vitamin eksikliği yada KC bozukluğundan kaynaklanan düşük düzeydeki multiple koagülasyon faktörleri; (3) kalıtımsal yada edinsel tek bir faktör eksikliği; (4) dissemine intravasküler koagulasyon (DIC) gibi tüketim koagulopatileri; ve (5) faktör VIII’e karşı gelişen antikorlar gibi koagülasyon faktörlerine karşı dolaşımdaki inhibitörler. Ek olarak; kan damarlarından kaynaklanan intrensek bozukluklar da kanama diatezine yol açabilirler. Laboratuvar değerlendirmelerinin, bu kategoriler göz önüne alınarak yapılmaları daha etkili sonuçlar verecektir.
Vasküler purpura (bruising) kan damarlarının intrensek yapısal anormalliği ile ya da kan damarlarımn enflamatuar infiltrasyonu (vasculilis) ile oluşan kanama olarak tanımlanır. Her ne kadar vasküler purpura genellikle normal trombosit sayısı ve normal koagulasyon durumlarında kanamaya sebep olsa da; vaskulit ve damar hasarı trombositler ile koagülasyon faktörlerirıin sekonder tüketimine yol açacak kadar ciddi olabilir. Yaşlı kişilerde, sıklıkla kan damarlarını örten subkutanöz dokularda bozukluklar gözlenir ve bunlar senil purpura olarak adlandırılır; kan damarlarının hassaslaşmasına yol açan benzer deri bulguları steroid tedavisinin de sık etkilerinden birisidir. Bu bağlamda, kollagenin bozulması ve subkutanöz dokuların incelmesi, atrofi sonucu olarak vasküler purpuraya yol açar. Vasküler purpuranın diğer bir edinilmiş sebebi skorbüt ya da C-vitamini eksikliğidir. Skorbüt bireyin saç telleri etrafındaki kanamalar (perifolikuler hemoraji) ve kıvucık şekildeki saçlar ile karakterizedir. Vasküler purpura, klasik olarak üst uyluklarda eyer şeklinde yerleşim gösterir. Skorbütte ağızda görülen kanama odakları ise subkutanöz doku hasarına değil, gingivite bağlıdır. Bununla birlikte, skorbütlü bazı hastalarda ağızda kanama olmayabilir ve skorbüt bu açıdan dışlanamaz. Damar duvarının konjenital defektleri de vasküler purpuraya yol açabilir. Bu nadir sendromlardan pseudoksanthoma elasticum vasküler yapılardaki elastik fiberlerde defekt sonucu oluşan şiddetli Gl ve genitoüriner kanama ile ilişkilidir. Ehlers-Danlos sendromu da hem kan damarları hem de subkutanöz dokuda anormal kollajen molekülleri ile karakterizedir. Her iki sendromda da deride vasküler purpura görülmekle birlikte sadece pseudoksantoma elastikumlu hastalarda belirgin Gl kanama gelişir. Gl kanama ile ilişkili diğer bir kalıtımsal damar duvar defekti de herediter hemorajik telenjektazi’dir (Osler-Weber-Rendu Sendromu). Bu bozukluk; dudak, Gl sistem ve diğer mukoz membranlarda kan kabarcıklarını andıran anjıomatöz lezyonlarla sonuçlanan kan damar duvarı dejenerasyonu ile karakterizedir. Bu lezyonların hasarı ile kanama ortaya çıkma sıklığı yaş ile artmaktadır ve Gl lezyonlar sıklıkla demir eksikliği ile sonuçlanan belirgin kronik kanarnalara neden olmaktadır. Döküntü ve ateşle birlikte görülen ani başlangıçlı palpable purpura (derideki lokalize elle hissedilen hemorajiler) aseptik veya septik vaskülite bağlı olabilir. Septik vaskülit; meningokoksemi ve diğer bakteriyel enfeksiyonlar tarafından oluşturulabilir, sıklıkla trombositopeni ve pıhtılaşma zamamnda uzama ile ilişkilidir. Çocuklarda ve genç erişkinlerde görülen aseptik vaskülitin bir nedeni olan Henoch- Schönlein purpurası; deri, Gl sistem ve böbreklerin vaskülitidir ve sıklıkla bağusak duvarına kanamamn yol açtığı abdominal ağn ile ilişkilidir. Bu sendrom, viral bir prodromal evre sonrası ortaya çıkabilir; IgA nefropatisine benzeyen renal histopatolojik özellikler ve serum IgA immun kompleksieri göz önüne alınarak bir Immunoglogulin- A (lgA) hipersensivite reaksiyonu olduğu söylenebilir. AIlopurinol benzeri ilaçlarla oluşan ilaç hipersensitivitesi de yoğun kutanöz purpura ortaya çıkarabilir.Vasküler bozukluklarda kanamanın tedavisi ileriye yönelik olmalıdır. Senil purpura ve steroid bağımlı purpurada genellikle tedaviye gerek yoktur. Skorbüt oral askorbik asit ile düzeltilir. Ehlers-Danlos Sendromu, herediter hemorajik telenjektazi ve pseudoksantoma elasticum gibi konjenital bozukluklarda; kanamaya eğilimi arttırıcı ilaçlardan (aspirin vb.) kaçınılır ve destek tedavisi (demir desteği vb.) uygulanır. Herediter hemorajik telenjektazi de sistemik östrojen uygulanması; nazal mukozanın squamöz metaplazisini induklemek suretiyle lezyonları travmadan koruyarak epitaksisi azaltır. Septik vaskülit tedavisinde, açık şekilde primer olarak uygun antibiyotik tedavisine odaklanılmalıdır; aseptik vaskülitte ise steroidler ve/veya immunsupresif ajanlar daha etkilidir. Vaskulit, trombosit ve koagülasyon faktörlerinde tüketime yol açacak kadar şiddetli ise (daha sonra DIC bölümünde tartışılacak), trombosit, kriyopresipitat ya da taze donmuş plazma transfuzyonu endikedir. Trombositopenide tanı koyma yaklaşımı, düşük trombosit sayısının nedeninin sınıflanması ile başlar: (i) azalmış trombosit yapımı (2) artmış trombosit sekastrasyonu (3) artmış periferik trombosit yıkım temel nedenlerdir. Azalmış trombosit yapımı ve periferal sekastrasyon (splenomegali vb.) ya da artmış yıkım (immun trombositopenik purpura vb.) arasındaki aynmda klasik tanı koydurucu test, kemik iliği megakaryositlerinin sayı ve morfolojik özelliklerinin değerlendirilmesidir. KırmJZl hücre retikulasit sayısına benzer şekilde, retiküle trombosit sayısı, kanda dolaşan genç trombositlerin invaziv olmayan bir ölçümünü verir. (artmJş RNA içerikleri yoluyla tanımlanır.) Retikule trombositler; trombosit kinetiğinin periferik kan indeksi olarak kullanılmakta ve trombositopeni değerlendirmesinin bir parçası olarak görülmektedirler.
Trombositopeni (trombosit sayısl<150,000mcL) hastanede yatan hastalarda en sık karşımıza çıkan problemlerden biri kemik iliğinde azalmış trombosit yapımı; kemik iliği aspiratları ve biyopsilerinde megakaryositlerin azalması ya da hiç bulunmaması ve retiküle trombositlere düşük oranda rastlanması ile karakterizedir. Normal megakarysitopoezin supresyonu şu durumlarda ortaya çıkar: (I) sitotoksik kemoterapi ile kemik iliği hasarı ve kök hücrelerin yıkımı; (2) normal ilik yapısının bozulması ve malign hastalık, apıazi, enfeksiyon (milier tüberküloz gibi) veya miyelofibrozis sonucu normal kök hücrelerin değişikliğe uğraması; (3) megakaryositik kök hücrelerin spesifik intrensek defektieri; ve (4) megakaryosit maturasyonunu etkileyen metabolik anormaliteler. Trombositopeni; malign veya otoimmun hastalıklar için uygulanan sitotoksik ya da immunsupresif kemoterapiye bağlı oluşabilir. Trombositopeni sıklıkla geri dönüşümlüdür ve trombosit yapımı megakaryositik kök hücrelerin geri dönüş ve rejenerasyonu ile ilişkilidir. Bununla birlikte; tekrarlayan ve/veya yoğun kemoterapi (kök hücre transplantasyonu vb.) megakaryositik kök hücrelerde ve destek stromal yapılarda kalıcı hasara yol açarak kronik trombositopeni yaratabilir. Bu durum, refrakter anemiyi (miyelodisplazi) düşündüren lökopeni ve anemi ile ilişkili olabilir. Tiazid grubu diüretikler gibi sık kullanılan ilaçlar, alkol ve östrojen preparatları da kemik iliğinde megakaryosit hasarına yol açabilir. Özellikle alkolizm ve anormal folat ya da vit-B 12 metabolizması ile ilişkili beslenme bozuklukları da trombositopeniye neden olan faktörlerdir; trombosit sayıları alkol bırakılmasına ve uygun multivitamin replasman tedavilerine cevap verir. Trombosit yapımı; lösemi ve multiple miyelom gibi kemik iliğinin intrensek malign hastalıkları ve kemik iliğini sekonder olarak tutan malign hastalıklar (non-Hodgkin lenfoma, küçük hücreli akciğer kanseri, meme ve prostat kanseri vb. diğerleri) tarafından baskılanabilir.
Bu durumlarda kemik iliği aspiratında azalmış megakaryositler ve nadiren malign hücreler görülür; kemik iIiğinin malign tutulumunun tanısı için kemik iliği biyopsisi daha değerlidir. Kemik iliği aspiratında, klonal-B hücrelerin flow-sitometrik değerlendirilmesi, lenfoproliferatif hastalıkların tanısında yüksek oranda etkilidir. (non-Hodgkin lenfoma) Kemik iliğinin retikülin fiberlerinde (ve bazende kollajende) artışla belirgin Miyelofibro:is de trombositopeni veya pansitopeniye yol açabilir. Miyelofibrozis en sık olarak miyeloproliferatif bozukluklarda, mastositoziste, kemik iliğinin mikobakteriyel ve diğer enfektif hastalıklarında ortaya çıkar. Aynı zamanda nadiren miyelodisplazi veya akut lösemili hastalarda, özellikle megakaryositik FAB MTde (Fransız, Amerikan ve İngiliz lösemi sınıflama şemasına göre) ve daha nadir olarak konjenital nedenli (osteogenezis imperfecta) olarak görülebilir. Trombositopeni aynı zamanda şiddetli apıastik anemisi olan hastalarda da görülür ve kemik iliği megakaryosit sayısı etkilenen diğer hücre tipleriyle birlikte azalmış ya da tamamen tükenmiş durumdadır.
Çocuklardaki trombositopeni; trombositopeni-radius yokluğu sendromu, konjenital amegakaryositik trombositopeni (frombopoetin reseptörlerindeki mutasyona sekonder) ve Fankoni anemisi (renal hipoplazi ve deri hiperpigmentasyonuyla giden konjenital apıastik anemi) gibi megakaryosit yapımındaki konjenital defektIerden kaynaklanabilir. Kemik iliği ile ilişkili diğer intrensek bozukluklar; periferal kan örneklerinde dev trombositler ve Döhle cisimcikleri (lökosit ve trombositlerde bazofilik inklüzyonlar) ile karakterize May Hegglin Anoma/isi ve ilişkili miyozin 1Ia/MYH9 gen hastalığıdır. WiskOff- Aldrich sendromu, egzema ve immun eksiklikle giden X-bağlı bir hastalıktır; bunun yanı sıra flow sitometri ile lenfositlerde CD43 eksikliğinin gösterildiği küçük trombositler ile beraber trombositopeni tanı koydurucudur. Sinirlerde duyarsızlık ve nefrit ile beraber konjenital hipoproduktive trombositopeni de Alport sendromunun bir parçasıdır. Kökeni ne olursa olsun, özellikle de malign hastalık için kemoterapi tedavisi gören hastalarda, hipoproduktive trombositopeniyi desteklemek için trombosit transfüzyonu uygulanır. Trombositopenide profılaktik trombosit transfüzyonu uygulaması: kemoterapi alan hastalarda kanama hastalarına göre daha sıktır. Profilaktik transüzyon için sınır trombosit sayısı genellikle 10,000/mcL olarak kabul edilir; bu değer ateşi, sepsisi veya Gl kanaması olmayan göreceli komplike olmamış klinik tablodaki hastalar için uygun ve güvenli sınır değerdir. Sınır değerin 10,000/mcL olarak seçilmesi belirgin şekilde trombosit transfüzyon sıklığını azaltır. Şayet komplike durumlar mevcutsa yada hastalara belli bir işlemle girişim uygulanıyorsa, trombosit transfüzyonları 20,000/mcL. altında uygulanır. Artmış periferik trombosit yıkılımı için herhangi bir neden olmayan hastalarda, her bir ünite donor trombosit konsantrasyonu, trombosit sayısını yaklaşık 10,000/mcL. düzeyinde arttırır. Böylece; yaklaşık 10,000/mcL. düzeyinde trombosit sayısı olan bir hastaya altı ünite trombosit transfüze edildiğinde trombosit sayısı yaklaşık 70,000/mcL. düzeyine çıkar. Bunun yanı sıra, trombositopenili hastalarda birliktelik gösteren ateş, sepsis, alloimmunizasyon, amfoterisin- B kullanımı, graft-versus-host hastalığı yada DIC olması trombosit tüketimini arttırır ve trombosit sayısındaki yükselmesinin belirgin olmasını engeller. Alloimmunizasyonu hariç tutarsak; sayılan durumlar genellikle transfuze edilmiş trombosillerin vücutta kalım ömrünü azaltırlar, fakat pratikte bu durum farklıdır ve immediate platelet recovery (ani trombosit iyileşmesi) olarak adlandırılır. Bu suretle, trombosit sayısı transfuzyon sonrası i saat içinde belirgin şekilde yükselir ve arkasından, komplike edici faktör birlikteliği olmayan trombositopenili hastalardakinden daha ani düşüşlerle azalır. Zıt olarak, alloimmunize hastalarda transfüzyon sonrası ilk saatteki trombosit yükselmesi görülmez ya da mjnimal düzeydedir çünkü alloimmunize hastalar sıklıkla, trombosit cross-match yada lökosit-antijen match ile donor taramasına ihtiyaç duyarlar. Transfuze trombositlere cevabı maksimal düzeyde gerçekleştirebilrnek için 1 saatlik transfuzyon sonrası trombosit sayısı önemli bir testtir. Bu durumdaki hastalarda, trombosit yüzeyinde taşınan ABO belirleyicilerinin yol açtığı azalmayı minimale indirmek için, mümkünse, tipe özgü trombositlerin transfüzyonuna çalışılmalıdır. Rh antijenleri, zıt olarak, trombosit yüzeyinde bulunmaz; buna bağlı olarak da trombosit alloimmunizasyonunda etkileri yoktur.
Dolaşımdaki trombositlerin %30 kadarı normalde dalak içinde bulunur. Splenomegaliye yol açan durumlar, trombosit yıkımında artma ile sonuçlanır. Bu trombosit sekastrasyonu trombositopeniye yol açar ve trombosit sayısını 50,000 ile 100,000/mcL. arasındaki değerlere ve nadiren de daha düşük düzeylere indirir. Sekastrasyondan kaynaklanan trombositopeni, ilerlemiş karaciğer hastalıklarında, splenomegaliyle ilişkili myeloproliferatif hastalıklarda (kronik muyelojenoz lösemi, honik idiopatik miyelofibrozis vb.) ve dalağı tutan malign hastalıklarda sık görülür. Malign bozukluğu olan hastalarda splenektorru endike olabilir. Zıt olarak, splenektomi, portal hipertansiyondan kaynaklanan trombositoperrin tedavisinde nadiren kullanılır. Miyeloproliferatif sendromlu hastalarda splenektomi yapma karan bireysel olarak hastaya göre, hem cerrahi hem de hastalığa spesifik komplikasyonlar göz önünde bulundurularak verilmelidir.
Artmış periferal trombosit yıkımı (imrnun ya da nonimmun mekanizmalara bağlı) sıklıkla trombositopeniye yol açar. Otoimmun trombositopeni; sadece trombositlere yönelik primer bir immun bozukluk olabileceği gibi, sistemik lupus eritamatozus gibi diğer otoimmun hastalıkların sekonder komplikasyonu olarak da karşımıza çıkabilir. İmmun trombosit yıkımının patofizyolojik karakteristiği, dolaşımdaki poliklonal antitrombosit antikor düzeylerindeki artış ile ilişkilidir. Bu antikorlar genellikle trombosit membran glikoprotein reseplörlerine; en sık glikoprotein llb/Illa (GPIIb/Illa) ve daha az sıklıkla da GPlb’ye yöneliktir. Trombositlerin bu antikorlar ile kaplanması; retikuloendotelial sistem (RES) hücrelerindeki Fc reseplörlerince trombositlerin opsonizasyonuna yol açar. Antikor kaplı trombositler dalak, daha az oranda da karaciğer tarafından yıkılır. Bu bozukluklar genellikle kemik iliği megakaryosit sayısında artış ile belirlenen, kemik iliği trombosit yapımında belirgin artış gösterirler. Oluşan genç trombositlerde, artmış hemostatik fonksiyonla ilişkili olarak göreceli yüksek granül içeriği vardır. Trombosit yıkımını, azalmış trombosit üretirrunden ayırmada klasik yöntem, kemik iliğinin artmış ya da normal megakaryosit sayıları açısından değerlendirilmesidir. Bununa birlikte, daha önceden belirtildiği şekilde, dolaşımda retikule tromhosiılerin yüzdesinin artması özellikle immun bağımlı yıkım, trombositopeni ile ilişkilidir ve hatta trombosit yıkımı tanısı için yeterlidir. İmmun yıkımdan kaynaklanan trombositopeni şiddetli olabilir ve trombositlerin yıkım süresi 7- 10 günlük normal süreden 1 günün altına inebilir. 1000-2000 mcL. arasındaki şiddetli trombositopeniye rağmen, ciddi kanama veya hemorajik ölümler nadirdir; bu kısmen genç trombosit fonksiyonlarının artması kısmen de vasküler bütünlüğü sağlamak için gerekli olan dolaşımdaki trombosit sayısının günde sadece 7100/mcL. gibi göreceli düşük bir sayıda olması ile açıklanabilir.
Çocuklarda, akut lTP sıklıkla variceıla gibi bir viral enfeksiyon sonrası ortaya çıkar. lTP’li hastalarda; peteşial hemoraji, mukozal kanama ve sıklıkla 20,000/mcL. altında trombosit sayısıyla belirgin trombositopeni görülür. Periferik kan örneklerinde büyük trombositler görülürken, çocukluk lösemilerinde görülen blastlar gibi anormal hücreler görülmez. Kemik iliğinde artmış ya da nadiren normal sayıda megakaryosit görülür. lTP tanısı kısmen diğer faktörlerin dışlanması ile konur. Ateş, organomegali, pansitopeni, lenfadenopati ya da anormal periferal kan hücreleri bulunması durumunda; lösemi, nöroblastom, Wilms tümörü yada diğer kemik iliği hastalıkları gibi malign hastalıkların acilen değerlendirilmesi gerekir. Laboratuar testleri klinik değerlendirmeye yardımcı olmakla birlikte, lTP tanısı için kesin gerekli değildir. Bu testler; periferik kanda artmış oranda retikule trombositlerin gösterilmesini ve serumda ya da trombositlerde trombosit otoantikorların saptanmasını içerir. Bununla birlikte; trombosit bağımlı antikor ölçümü her ne kadar duyarlıolsa da, ITP’ye spesifik değildir; çünkü trombositlere nonspesifik bağlanan immunglobulinler trombositopenili hastalarda sıklıkla karaciğer hastalığı veya human immunodeficiency virus (HIV) enfeksiyonu gibi diğer nedenlere sekonder artış göstermektedir. Zıt olarak, spesifik trombosit glikoproteinlerine karşı serum antikorlarını ölçen teknikler daha fazla spesifiteye sahip olmakla birlikte, göreceli olarak duyarlılıkları düşüktür. Ortalama trombosit hacmindeki artış da, normal değerlerin geniş bir aralıkta seyretmesinden dolayı, yıkıma bağlı trombositopeninin göreceli olarak duyarlılığı düşük ve spesifik olmayan bir belirleyicisidir. Retikule trombosit yüzdesindeki artış, artmış trombosit yıkımı ile ilişkili olmakla birlikte; ITP ile heparin-bağımlı trombositopeni (HIT) ve trombotik trombositopenik purpura (TTP) gibi trombosit yıkımının diğer sebepleri arasında ayrım yapamaz. Bu açıdan ITP teşhisi halen büyük oranda klinik bulgulara dayanmaktadır. Çocuklarda akut TTPtedaviye gerek duyulmadan düzelebilir, fakat klinisyenlerin büyük çoğunluğu bu çocukları steroidler ya da intravenöz immunoglobulin (IVlG) ile tedavi etmeyi tercih etmektedirler. TTP’de kullanılan IVIG tedavisinin farklı mekanizmalarla etki gösterdiği düşünülmektedir: (i) yüksek immunoglobulin G (IgG) konsantrasyonları, RES fagositlerindeki Fc reseptörlerini ve antikor bağımlı sitotoksisite selluler efektörlerini bloke ederler; (2) IgG infüzyonu IgG katabolizması fraksiyonel yüzdesini arttınr ve bu suretle konsantrasyonla direkt orantılı olarak antitrombosit IgG yıkımını arttırır; (3) antitrombosit 19 temizlenmesi antiidiotipik etkilerle arttınlabilir. (ITP antikorlarına immunolojik cevap meydana getirmek vb.) Akut ITP’li çocukların %80’inden fazlasında hızlı bir remisyon vardır ve ITP tekrarlamaz. Bulguları düzelen hastaların %10-20’sinde ise rekurren trombositopeni (kronik ITP vb.) gelişir; bununla birlikte bu çocukların %70’den fazlası splenektomiye tam cevap verir. Kronik gidişi i bu vakalarda; splenektomi sonrası, epizodik IVIG, bu bölümde tartışılan) ve ciddi vakalarda immunsupresif tedavi kullanılmaktadır. Hemorajik ölümler çocukluk ITP’lerinde nadirdir «%2), fakat kronik, refrakter TTP ile ilişkili bazı mortalite oranları da vardır. (%2-%5) Çocuklarda olduğu gibi, erişkinlerdeki ITP teşhisi de büyük oranda dışlama ile yapılır; fakat çocuklardan farklı olarak, erişkinlerdeki akut ITP nadiren spontan düzelir ve büyük oranda kronikleşir, hastaların %50’den fazlasında kronik ITP gelişir. Peteşial hemoraji ve mukozal kanama 20,000/mcL altındaki trombosit seviyeleri, sıklıkla da 1000- 2000 mcL. kadar düşük düzeyler, ile ilişkilidir. ITP’li erişkinlerde %10’un altında hemorajik ölümler ortaya çıkar. Erişkinlerde ITP, HIVenfeksiyonu gibi diğer hastalıklar ile ilişkili olabilir. ITP, HIVenfeksiyonunun ilk bulgusu olarak karşımıza çıkabilir; oysaki HIVenfeksiyonunun ileri evrelerinde görülen trombositopeni daha sık olarak HIV ile megakaryosit enfeksiyonu sonucu kemik iliği yetmezliği, kemik iliğinin mikobakterial enfeksiyonu ve son dönem HIV hastalarındaki beslenme bozuklukları ile ilişkilidir. ITP aynı zamanda sistemik lupus eritematozus, inflamatuar barsak hastalığı ve hepatit gibi otoimmun bozuklukları olan hastalarda da görülür. De novo ITP’de olduğu gibi artmış periferik trombosit yıkımı, kemik iliği incelemesinde normal veya artmış megakaryosit sayısı, bu otoimmun hastalıklarda trombositopeniye neden olur. Bazı hastalarda ITP’ye otoimmun hemolitik anemi eşlik eder; bu durum Evans sendromu olarak adlandırılır. Eritrositler için Cooms testi genellikle sıcak-reaktif otoantikorlarla pozitiftir. Başlangıç tedavisi sadece ITP olan hastalarda belirgin bir şekilde farklı değildir fakat kronik Evans sendromunun, tek başına ITP’li hastalara göre, genellikle splenektomiye daha az cevap verdiği düşünülmektedir.
Sistemik lupus eritematozus zemininde ITP; trombosit yüzeyinde immun kompleks birikimi ve aktif vaskülit gibi trombosit yıkımını arttırarak trombosit sayısını düşüren, otoimmun hastalığın kendisi ile ilişkili faktörlere bağlı olabilir. Hem ITP, hem de altta yatan otoimmun hastalığın tedavisi genellikle beraber yapılır. Sistemik lupus eritematozus ve trombositopeni ile ilişkili olarak lupus antikoagulant veya antikardiolipin antikorlar varsa, seconder antifosfolipid antikor sendromu tanısı konur; bu durum sıklıkla tromboembolik komplikasyonlar ile ilişkilidir. İmmun trombosit yıkımı, aynı zamanda ilaçlar ile ilişkili olabilir. Kinidin yada kinidin türevi ilaçlar trombositlere bağlanırlar ve trombosit ile ilacın birlikteliği hapten denilen yeni bir antijen oluşturur. Antikorlar bu yeni antijene yönelirler ve RES tarafından trombositlerin hızla yıkımına neden olurlar. Trombositopeni gelişimi genellikle hızlıdır ve ilaç maruziyetine bağlı geçici bir durumdur; ilacın kesilmesi ile trombosit sayısı aynı hızda normale döner. ITP’ye yol açan diğer ilaçlar arasında sulfa bileşikleri, altın tuzları ve psikotropik ilaçlar sayılabilir. İlaca ara vermek her zaman gereklidir, aynı zamanda bazı durumlarda steroid ya da IVIG tedavisine de gerek duyulabilir. Erişkinlerde akut ITP tedavisinde ilk seçilecek ilaç steroidler,1-2 mg/kg/gün dozunda prednizon’dur. Trombosit transfuzyonu, genellikle ITP’de tercih edilmez çünkü transfüze trombositlerin yaşam süresi kısadır ve kanama komplikasyonları seyrektir. Bununla birlikte, belirgin kanaması olan veya cerrahi girişim gereken hastalarda, trombosit transfüzyonları güvenli olarak yapılabilir ve 24 saatten az kısa bir sürede trombosit sayısında yükselme sağlanabilir. Şiddetli trombositopenisi veya hayatı tehdit eden edici kanaması olan akut ITP’li hastalarda, yüksek doz metilprednizolon (1gr/gün 3 gün) tek başına veya IVlG (bölünmüş dozlar şeklinde 2g/kg, 2-5 gün süreyle) ile kombine edilerek kullanılabilir. Rekürren ITP’de, kronik steroid tedavisi sıklıkla gereklidir fakat genellikle belirgin yan etkiler gösterir. Bulgular göstermektedir ki kronik ITP’li hastalar, gerek çocuk gerekse erişkin, başlangıçta IVIG tedavisine cevap verirlerse, arkasından gelecek splenektomiye de iyi cevap verirler; oysaki IVIG tedavisine cevap vermeyenlerde splenektomi sonrası hastalık remisyonu da daha nadirdir. Her ne kadar, splenektomi yapılan hastaların yaklaşık 1/3’ünde kronik ITP devam etse de, kronik ITP’li hastaların %50’den fazlasında splenektomi sonrası bir dereceye kadar hastalık remisyon göstermektedir. Splenektomi sonrası şayet ITP düzelmezse KC ve dalak taraması ile aksesuar dalak varlığı dışlanmalıdır çünkü Howell-Iolly cisimleri halen bulunabilir. Rekurren hastalık sıklıkla epizodiktir, özellikle viral enfeksiyonlar sonrası görülür ve bu hastalarIVlG ile ya da Rh(+) olanlar RhoGAM ile tedavi edilebilir. RhoGAM, grup Rh D antijenli kanlardaki antikordur, RES’de Fc reseptör blokajı ve karaciğerdalak trombosit up-take’ini azaltarak kırmızı hücre hemolizini sıklıkla hafif oranda artırır. Özellikle HIV enfeksiyonlu olan ITP hastalarında Rhogam sonrası belirgin hemoliz oluşmaktadır ve bu tedavi dikkatle takip edilmelidir. Rhogam splenektomi geçirmiş hastalarda etkisizdir. Splenektomiye cevap vermeyen hastalarda steroid dozu, danazol, kolşisin ya da siklofosfamid gibi immünsüpresiflerin eklenmesiyle güçlendirilir. Kronik ITP’li bazı hastaların anti-CD20 monoklonal antikor infıizyonuna cevap verdikleri bilinmektedir. ITP’li erişkinJerin yaklaşık %5’i kronik refrakter hastalıktan dolayı ölmektedir.
Anne, sık olmayan GPIIIa üzerinde PI(A2) (heparin bağımlı antikor [HPA]- ib) gibi trombosit alloantijenlerine homozigot olduğunda ve fetüs babadan iletilen PI(AI) (HPA-I a) haplotipi gösterdiğinde; neonatal alloimmün trombositopeni ortaya çıkar. Aııoimmün trombositopeni patogenezi, Rh sensitizasyonunun ortaya çıkardığı yenidoğanın hemolitik hastalığı ile benzerdir. İlk gebelikte P 1(A1) antijenine maruz kalan anne ikinci ve sonraki gebeliklerde PI(AI)'e karşı yüksek titrede IgG antikorları oluşturur. Bu antikorlar plasentayı geçerler, PI(AI) fetal trombösitlerle reaksiyona girerler ve RES 'de periferik trombosit yıkımına yol açarlar. Neonatal alloimmün trombositopeni şiddetli olabilir, ancak bu reaksiyon kanamanın uterusta mı, doğum sırasında mı yoksa hayatın ilk günlerinde mi olacağını tahmin etmemizi sağlayamaz. Kanamayı tedavi etmek ve trombosit sayısını sabitlemek için IVIG ve PI(A!) içermeyen anne trombositleri kullanılır. Alloimmün trombositopeni transfüzyon sonrası erişkinlerde de görülebilir. (Posttransfüzyon purpura) Neonatallerde olduğu gibi bu durum hastanın normal trombositlerinde bulunmayan P i(A i) gibi sık görülen trombosit alloantijenlerine maruziyete bağlıdır. Bu bozukluk sıklıkla kırmızı kan hücresi ya da trombosit transfüzyonu sonrası PI (A2) için homozigot olup önceki gebelik sonucu PI(Al) ile aııoimmünize olmuş kadınlarda; veya daha nadir olarak önceki transfüzyonlara bağlı alloimmünize herhangi bir hastada görülür. Kan donörlerinin %90’dan fazlası PI (A i) içerir ve P i(Al) trombositler tarafından sunulur. Sonuç olarak az oranda trombosit kontaminasyonu içeren kırmızı kan hücre ürünleri ile PL (A i) içerir. Kan ürünlerine cevap anamnezi rezidüel donör trombositlerinde ve hatta daha ilginç olarak Pl(Al) allaantijen içermeyen doğal trombositlerde bile yıkım ile sonuçlanır. Her ne kadar bulgular, normal trombositlerin nonspesifik olarak RES tarafından ya da trombositlere PI(AI) adsorbsiyonu ile yıkıldığın! gösterse de, posttransfüzyon purpuranın patofizyolojik yönü (PTP) açık değildir. Neonatalerde olduğu gibi bu hastalar IVIG ile tedavi edilirler ve transfüzyonlar homozigot PI(A2) donörlerinden türetilmelidir. Her ne kadar alloimmün trombositopenin en sık nedeni PI(A2) ise de bu klinik sendroma yol açan başka trombosit aııoantijenleri de vardır. Neonatallerdeki trombositopeni aynı zamanda maternal ITP’ye de bağlı olabilir. Antitrombosit antikorlar, sıklıkla 19 G antikorları plasentayı geçerek fetüsta trombositopeni oluşturabilider. Her ne kadar bazı bulgular annede ITP bulunduğunda ve anne trombosit sayısının 75000/mcL. altında olduğunda neonatal trombositopeni insidansının attığını gösterse de; anne ITP’siyle birlikte görülen belirgin neonatal trombositopeni nadirdir ve risk altındakilerin % lO’dan azında görülür. Bazen annedeki otoantikorların plasental geçişini azaltmak amacıyla anneye ITP tedavisi uygulamak gerekir, bununla birlikte maternal ITP durumlarının çoğunda fetal trombositopeni sık görülmez ve güvenli vajinal doğum gerçekleştirilebilir.
Her ne kadar immün doğada olsa da potansiyel yıkıcı frombofik komplikasyonları ve patofizyolojik özelliklerinden dolayı HIT diğer ilaç bağımlı ITP formlarından ayrılmalıdır. UFH maruziyeti olan hastaların %25’inde heparin ve trombosit aktivasyonu sonrası alfa granüllerden salınan trombosit faktör iV (PF IV) kompleksini tanıyan antikorlar gelişir (enzim bağımlı immünosorbent assay) [ELISA]. Sıklıkla trombosit sayısı 50000-100000/mcL arasında olan bu hastalar tekrardan heparin aldıklarında % ıo ila 20 HIT gelişir. Cerrahi girişim HlT için spesifik bir risk faktörüdür; kardiyopulmoner bypassnedeniyle kardiyak cerrahi uygulanacak hastalarda, kalça replasmanı yapılacak hastalara göre daha yüksek oranda HIT antikorları ortaya çıkar. UFH’ye zıt olarak sadece düşük moleküler ağırlıklı heparin (LMWH) alanlarda HIT insidansı, UFH hastalarıyla kıyaslandığında 1/5-1/10 oranında daha düşüktür. Bununla birlikte hem UFH hem de LMWH için trombositopeni mekanizmalarıbenzer görülmektedir: Trombosit Pc-reseptörlerine heparin PP IV antikor kompleksi bağlanması sinyal iletimini sağlayarak trombosit aktivasyon granül salınımını indükler ve trombosit yüzeyinde trombin oluşumunu artırır. Aktivasyon ile trombositlerin yıkımı trombositopeni ile sonuçlanır. Teşhis predominant olarak klinik ile yapılır, fakat hızlı ELİsA testiyle de heparin-PP IV antikorları saptanabilir. ELİSA’nın eksik yönü antikor kompleksinin trombositlerin fonksiyonel aktivatörü olup olmadığını saptayamamasıdır; bu açıdan HIT için duyarlı fakat özgül olmayan bir testtir. Serotonin salınım testi, heparin varlığında serum antikor heparin maruziyeti sonrası trombosit aktivasyonu saptanmasında HIT için fonksiyonel bir testtir. HIT’de trombin bağlı prokoagülan cevap trombositleri yok eder ve hastaların %10-20’den fazlasında şiddetli ya da hayatı tehdit edici trombotik komplikasyonlara yol açar. Her ne kadar HIT ile birliktelik gösteren kardiyovasküler hastalığı olan ve tam doz heparin alanlarda, trombozis sık görülse de herhangi bir heparin dozunda (heparin damlacıklarında bile) HIT’de trombozis oluşabilir. Hasta heparin alıyorken ve hatta heparin kesildikten haftalar sonra, büyük olasılıkla dolaşımda kalan prokoagülan trombosit mikro partikülleri ile ilişkili olarak arteriyal ve venöz tromboemboli oluşabilir. Her ne kadar antikodar UPH tedavisiyle indüklense de bu antikorların %80’den fazlası LMWH ile çapraz reaksiyon gösterir ve yaklaşık %15’i de heparinoid ile reaksiyona girer; bu açıdan heparine son verilmesi kritiktir. Buna bağlı olarak HIT’li hastalarda kısa dönem antikoagülan olarak tercih edilen tedavi; lepirudin ve argatroban gibi direk trombin inhibitörleridir. Heparin-PP IV antikorları bu bileşiklerle reaksiyon vermez; gerekli görüldüğünde uzun dönem antikoagülasyon için, trombosit sayısı normale geldiğinde warfarin eklenebilir. HIT’li hastalarda kısa dönem tedavide, direk trombin inhibitör koruyucusu olmadan warfarin kullanılmamalıdır. HIT’li hastalarda tek başına warfarin kullanımı; büyük olasılıkla warfarin deri nekroz sendromuna (bölüm 53) benzer şekilde, edinilmiş protein C eksikliğiyle ilişkili olarak uzuvlarda ciddi trombozise yol açar. Non-immün periferik trombosit yıkımının sık görülen ve potansiyel hayatı tehdit edici nedenlerinden biri olan Dİc sepsis, malignite, ileri karaciğer hastalığı ve diğer rahatsızlıkların yol açtığı endotoksin salınımı sonucu oluşan ciddi doku hasarı ile ilişkilidir. Bakteriyel sepsisin yol açtığı DİC’te dolaşımdaki endotoksinler, dolaşımdaki monositler ve endotelya! hücrelerdeki doku faktörlerinin tanımlanmasını artırırlar; bu işlem trombin ve fibrin oluşumunu etkiler. Pibrinin, fibrinolizle göreceli olarak uyumlu şekilde vasküler yapılarda birikimi; trombotik ya da mikroanjiopatik vaskülopatiye ve sonrasında takip eden organ hasarına yol açar. Trombositlerin ve dolaşımdaki faktörlerin trombin ile aktivasyonu sonucunda kemik iliği ve karaciğerin sentetik kapasitesi göreceli olarak etkilenir; bu da PT ve PTT sürelerinde uzama sonucu trombositopeni ile sonuçlanır. DİC’te primer lezyon her ne kadar pıhtı oluşumuysa da klinik son nokta genellikle tüketim koagülopatisidir. Tüketim koagülopatisinden kaynaklanan trombosito-peni ve düşük faktör düzeyleri özellikle gastrointestinal sistemde intravenöz ek yerlerinden görülen karakteristik sızıntı şeklinde mukozal kanamalar ile belirgindir.
DİC’in tüketim koagülopatisinde fibrinojen düzeyleri sıklıkla düşüktür, fakat normal ya da hafif yüksek de olabilir; sepsis veya altta yatan bozukluğa akut faz reaksiyonu fibrinojen salınımını artırabilir ve DİC’in ortasında normal düzeylere getirebilir. Bu açıdan şayet fibrinojen normal düzeylerdeyse Dİc dışlanmamalıdır. DİC’teki fibrinoliz fibrin pıhtısı ve doku tipi plazminojen aktivatörleri tarafından tetiklenir; laboratuar tetkikleri genellikle 40 mcg/mL üzerinde fibrin yıkım ürünü (fibrin monomer parçaları) ve 0.5 mcg/mL üzerinde Ddimerler (firin-fibrin bileşik parçaları) düzeylerindeki artışı gösterir. Her ne kadar fibrin yıkım ürünleri DİC’li hastalarda yüksekse de bu bulgu spesifik değildir; zıt olarak yüksek D-dimer düzeyleri Dİc için daha spesifiktir ve sıklıkla tetkiklerdeki fibrin yıkım ürünlerinin yüksek düzeylerinin doğrulanması için kullanılır. Periferik kan örneklerinde belirgin sayıda şistositlerin görülmesi de mikroanjiopatik tablonun teşhisine yardımcı olabilir; bununla birlikte bu bulgu DİC’e spesifik değildir ve aynı zamanda TTP durumlarında da görülür (ileride anlatıldığı gibi). Kronik Dic; anevrizma, hemanjiyom ve mural trombüslerle ilişkili olarak trombosit ve faktörlerin büyük pıhtılarca tüketimi sonucu oluşabilir. Kronik DlC’in özel bir sebebi de sıklıkla adenokarsinom ya da akut promiyelositik lösemi gibi malign hastalılardır; bu bozukluklardaki malign hücreleraktif faktör X veya Faktör Xa aktivitesini artıran maddeler salgılarlar. Xa aktivasyonu protrombin kompleksi oluşumuna, trombin yapınlina ve trombosit aktivasyonu na yol açar; bu durumda kronik DIC, genellikle PT ve PTI’de belirgin uzamayla karakterize faktör tüketimine yol açar. Klinik olarak bu hastalarda gezici tromboflebit (trousseu sendromu) veya non-bakteriyal (marantik) endokardit görülür. Dic tedavisi şunları amaçlamalıdır: (l) Sepsis için antibiyotik uygulanması veya malign hastalıklarda kemoterapi gibi altta yatan hastalığın tedavisi; (2) trombositler, kriyopresipitat (fibrinojen için), ve taze donmuş plazma gibi destekleyici hemostatik tedavi; (3) Koagülasyon faktörleri ve trombositlerin aktivasyonunun bozulması. Bu son yaklaşım için; prokoagülana karşı antikoagülan aktivite dengesi pıhtı oluşumu yönünde aktif olarak bozulmadıkça, mural trombüs ile birlikte görülen arteriyal tromboemboli veya Trousseau’s sendromuyla birlikte görülen gezici tromboflebitteki gibi; genellikle antikoagülasyon endike değildir. Kronik DİCteki bu trombotik komplikasyonlar sıklıkla varfarin tedavisine dirençlidir; Dİc rezolüsyonu genellikle yoğun anti-Xa tedavisi (UFH ya da LMWH) yanı sıra altta yatan malign hastalık ya da tüketim bozukluğunun başarılı tedavisiyle mümkündür. Anti- Xa ajanlara ek olarak sepsis ve DİCe karşı yeni tedavilerin de etkinliği gösterilmiştir; farmokolojik aktive protein Cnin sepsis ve/veya DiCte mortaliteyi belirgin azalttığı gösterilmiştir ve DİCteki koagülasyon işlemini düzenlemeye yönelik diğer metotlar araştırılmaktadır

Trombosit aktivasyonu ve yılanundan sonuçlanan diğer bir non-immün trombositopeni sebebi de TIP’dir. Konjenital teharlayan TIP’li hastalarda ADAMTS 13 olarak bilinen vWF-cleavig proteaz belirgin olarak azalmış veya hiç yoktur. Aile hikayesi olmayan edinilmiş TTP’li hastalarda sıklıkla IgG yapısında vWF cleavig proteazın normal fonksiyonlarını bloke eden bir antikor vardır. Proteaz fonksiyonlarında eksiklik azalmış yıkıma ve yüksek düzeyde yüksek moleküler ağırlıklı vWF multimerlerinin dolaşımına yol açar; bu durum koagülasyon mekanizmasında aktivasyon olmadan artmış trombosit adezyonu ve yıkımı ile sonuçlanır. Bu açıdan hem PT hem de PTI normaldir kemoterapi ve gebelik ya da HIV enfeksiyonu ile ilişkili durumlar sonrası oluşan TIp’de de benzer patogenez görülmektedir. Sıklıkla şiddetli trombositopeni periferik kanda şistositlerle birlikte görülen mikroanjiopati ve artmış serum laktat dehidrogenaz ile ilişkilidir. Değişik organlarda, özellikle böbrek ve beyinde, görülen mikrovasküler oklüzyonlar çok değişik semptomlara yol açarlar. Klasik beş bulgu (ateş, trombositopeni, mikroanjiopatik hemoliz, nörolojik semptomlar ve renal yetmezlik) TTP’li hastaların %25’inden azında görülür. ADAMTS 13 aktivite ve inhibitörlerine yönelik testlerin kullanımı büyük oranda halen mümkün olmadığından ve testlerden hızlı cevap alınamadığından; teşhis genellikle klinik tanı ile yapılır. TTP tedavisi, antikorların yok edilmesine ve bağlanan proteaz aktivitesinin yenilenmesine dayanır. Bu amaca genellikle plazma değişimi ile ulaşılır, hasta plazması taze donmuş plazma ile değiştirilir (plazmaferez), transfuze plazmadaki vWF multimer düzeylerini azaltmak için sıklıkla ‘cryo-poor’ uygulanır. Aynı anda sıklıkla steroidler ve antitrombosit ilaçlar (aspirin, dipridamol vb.) da verilir, fakat her iki ilacında göreceli faydaları tam net değildir. TTP’de trombosit transfüzyonları da göreceli olarak kontrendikedir; fakat invaziv işlemlerden önce kullanıldığında yan etki olmamaktadır. TTP tedavisinde, rekombinant ve/veya purifiye formlardaki ADAMTS13 de akılda bulunması gereken potansiyel terapötik ajanlardır. Otoritelerin çoğu; hemolitik üremik sendromun (HÜS), TTP spekturumunun bir parçası olduğunu düşünürler; bununla birlikte, HÜS’de görülen hemolitik anemi ve renal yetmezlik genellikle nörolojik yıkıınla birlikte değildir ve HÜS genellikle TIP ile aynı derecede trombositopeni ya da şistositozis göstermez. Ek olarak, HÜS bozulmuş vWFcleaving proteaz aktivitesi ile ilişkili değildir. TIP’den farklı olarak, HÜS genellikle çocuklarda görülür ve daha az sıklıkla da Shiga benzeri toksin oluşturan bakterilerin, özellikle Escherichia coli 0157.H7 serotip, yol açtığı hemorajik kolit şeklinde erişkinlerde görülebilir. Mikrovasküler trombosit trombuslerinin benzer patofizyolojik özellikleri; HÜS’un, TTP’nin bir devamlılığı olduğunu ve gerçekten HUS’lu hastalarda renal fonksiyonlar düzelinceye kadar devam edilecek diyalizin yanı sıra, plazma değişimi ile birlikte plazmafereze cevap verdiklerini düşündürmektedir.
Gebe kadınlarda, henrodilusyon ve gebeliğin normal fizyolojik durumu ile ilişkili olarak trombosit sayısı sıklıkla 100000-150000/mcL. aralığında olabilir; bu değerler maternal ya da fetal komplikasyonlar ile ilişkili değildir. Zıt olarak, platelet yıkımının otoimmun sebeplerinde (daha önceden bahsedilen) gebelik bağımlı hipertansiyon durumlarında platelet sayılan 100000/mcL. altına inerek komplikasyonlar ortaya çıkabilir. Cebelik bağımli hipertansiyon spekturumu; hipertansiyon ile, proteinüri ve böbreğin işlev bozukluğu (preeklempsi) ya da serebral ödem ve bayılmalar (eklempsi) arasında değişen geniş bir spekturumdur. Trombositopeni, sıklıkla doğum sırasında ya da üçüncü trimestrın ileri dönemlerinde, gebelik bağımlı hipertansiyonun bir geç bulgusu olarak ortaya çıkabilir. Gebelikle ilişkili HELLP sendromu hemoliz, karaciğer enzimlerinde yükselme, preeklempsiyle birlikte düşük trombosit sayısı ile karakterizedir. Gebelik bağımlı hipertansiyon ve HELLP ile ilişkili trombositopeni; büyük olasılıkla trombositlerin tüketimi, vaskulopati ve mıkrovasküler okluzyonlara yol açan anormal vasküler prostaglandin metaboLizması tarafından ortaya çıkarılmaktadır. Bu bozukluklar sıklıkla, fetus ve plasentanın doğumu ile geri dönüşıüdür. Zaman zaman, hastalığın tedavisinde IVIG ya da plazmaferez gerekebilir. Doğum sonrası trombositopeni düzelmezse, TTP gibi diğer alternatifler farklı durumlarda akılda tutulmalıdır.
ITP ile ilişkili sistemik lupus eritematozustan farklı olarak, antifosfolipid sendrom kanama ile ilişkili değildir. Antifosfolipid sendromunda; yıkıcı trombositopeni, tekrarlayan trombosis veya fetus kaybı, lupus antikoagulanların ve/veya antikardiolipin antikorları saptanması ile tanı konulur. Sistemik lupus eritematozus tanı hiterleri göstermeyen antifosfolipid sendrom, primer bir hastalıkolabilir yada gerçek sistemik lupus eritematozusa sekonder ortaya çıkabilir. Antifosfolipid sendromdaki trombositopeni, artmış periferal trombosit yıkımı sonucudur ve trombosit spesifik antikorlara bağlı değildir; mikrovasküler yapılarda vasküler anjiopati ve artmış trombosit tüketimi nedendir. Warfarin ya da LMWH ile uzun dönem yoğun antikoagülasyon, bazen aspirin veya diğer antitrombosit ilaçların eklenmesi, trombotik komplikasyonları önleyebilir ve trombosit sayılarını normal düzeye getirebilir.
Sekastrasyon, yapımda azalma ve trombositopeninin yıkıcı nedenlerinin yanı sıra; trombositopeni hemodilusyondan da kaynaklanabilir. Bu durum genellikle, travma ya da kardiyopulmoner bypass için massif kırmızı kan hücreleri ve plazma transfuzyonunu takiben; normal dolaşım sistemine ekstra yük eklenmesiyle ortaya çıkan belirgin hemodilüsyon durumlarında görülür. Bunun yanı sıra; bypass ın yarattığı hemodilusyona ek olarak, kardiyopulmoner bypassa maruz kalan dolaşımdaki trombositler, membran reseptör kaybı ve aktivasyonu ile, geçici olarak disfonksiyonel hale gelirler; bu defekt hafif ya da geçici olabilir, fakat özellikle uzun bypass işlemleri sonrası bazen şiddetli kanamaya yol açabilir. Bypassın tamamlanması yada akut travmanın çözülmesi sonrası, trombosit sayısı 48-72 saatte iki katına çıkar; bunun yanı sıra trombosit sayısı düzelirken şiddetli kanarnaları tedavi etmek için trombosit transfüzyonlarına ihtiyaç duyulabilir.
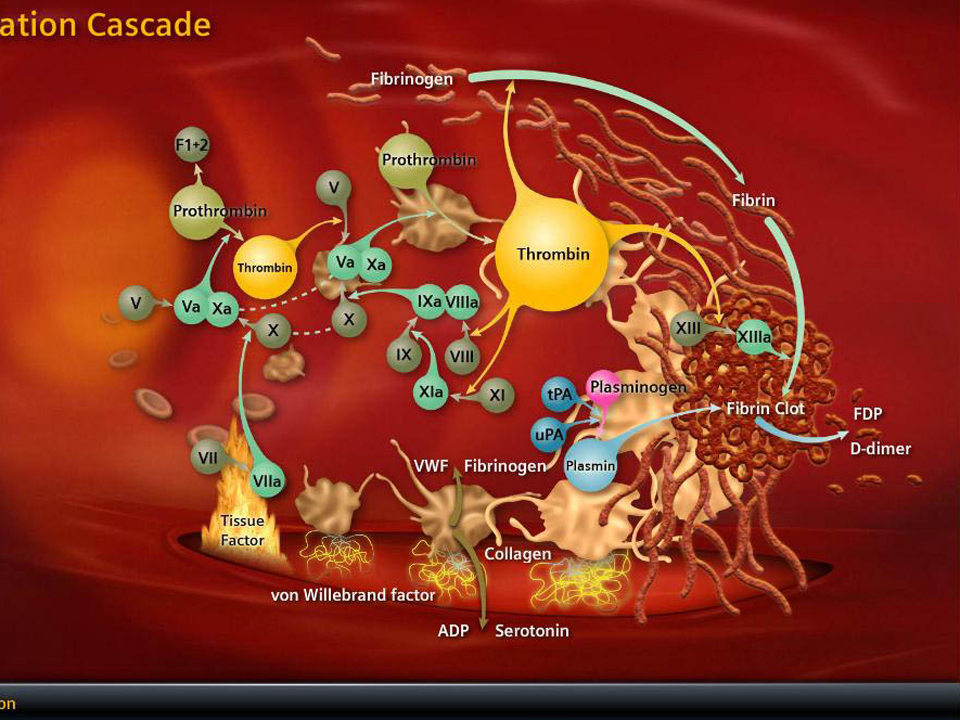
Özellikle cerrahi girişim uygulanacak hastalarda, primer hemostaz açısından, trombositlerin hasarlı damarlara yapışması ve pıhtı içine diğer trombositleri çekmesi kritiktir. Preoperative taramalarda önemli bir soru, hastaların aspirin gibi trombosit fonksiyonlarıyla etkileşim gösterebilecek ilaçları kullanıp kullanmadıklarıdır. Maruz kalan bütün trombositler geri dönüşümsüz olarak etkilenirler ve aspirin kesilse bile araşidonik asite cevap vermezler. Karakteristik aspirin bağımlı trombosit aggregasyon şekli gösterilmiştir. Zıt olarak; diğer nonsteroid anti-enflamatuar ilaçlar (NSAID) (indometazin vb.) siklooksijenazı (COX) geri dönüşebilir şekilde inhibe ederler, ve trombosit fonksiyonları ilaç kesilmesinden 24-48 saat sonra normale döner. Aspirin ve NSAI ile ilişkili kanamalar sıklıkla hafiftir ve özellikle inme ya da miyokard enfarktüsü riski olan hastalarda aspirin bağımlı trombosit işlev bozukluğu istenen bir şey olduğundan, aspirinin kesilmesini gerekmeyebilir. Bununla birlikte, aspirinin yol açtığı tedavi gerektiren kanamalarda desmopressin asetat (DDAYP) infüzyonunun, kanama zamanını azaltmada etkili olduğu gösterilmiştir; bazı durumlarda trombositlerin infuzyonu uygun olabilir. Yakaların çoğunda, dört ile altı randomize donörle yapılan tek bir trombosit transfuzyonu, primer hemostazı sağlayacak (total dolaşımdaki sayıdan >%10) yeterli normal trombosit düzeyini oluşturur. Benzer şekilde diğer ilaçlar tarafından oluşturulan trombosit işlev bozukluğunu ve kanama durumlarında da, ilacın kesilmesi ve trombosit transfuzyonu gerekebilir. Aspirinin etkisi COX- 1 ile sınırlıyken, farklı NSAID’lar COX-l ve COX-2 için göreceli olarak farklı afinitelere sahiptirler. COX-2; enflamatuar sitokin cevabında, endotel hücrelerde (trombositlerde değil) sentezlenen induklenmiş bir enzimdir. COX-2 supresyonu, prostaglandin 1.2 (prostasiklin, prostasiklin [PGI.2]) azalması ile sonuçlanır; prostaglandin 1.2, diğer fonksiyonlarının yanı sıra trombosit agregasyonu inhibisyonu ile antitrombotik etki gösteren bir maddedir. NSAİD’lerin protrombin ve/veya antitrombotik denge üzerindeki etkileri kanamayı oluşturur çünkü NSAID-bağımlı COX- 1 inhibisyonu, trombositlerde tromboxane A.2 (TxA.2) yapımını bloke eder. Zıt olarak; daha selektif COX-2 inhibitörleriyle birlikte görülen kardiyovasküler riskteki artma; trombosit fonksiyonlarının tam olmasıyla birlikte (TxA.2’de COX-2 blokajı ile inhibisyonolmaması), COX-2 bağımlı endotel hücre PGL2 yapımının gerçekleşmemesi ile ilişkilidir. Son veriler aynı zamanda göstermektedir ki; aspirinden önce verilen NSAID’ler COX-2 bağlanma bölgeleri için yarışırlar ve aspirinin antitrombositik etkisini azaltırlar, bu da NSAID kullanımının prokoagulan dengedeki diğer bir olası faktörüdür. Üremik trombasit işlev bozukluğu böbrek yetmezliğinde biriken ve vasküler endotel hücrelerde yüksek düzeyde nitrik oksit oluşumunu arttıran guanidinosuksinik asit (GSA) gibi proteinler tarafından oluşturulur. Her iki bileşikte trombosit fonksiyonlarını inhibe etmektedir, fakat veriler göstermektedir ki trombosit fonksiyonlan üzerinde GSA inhibitör etkisine nitrik oksit aracılık etmektedir. Renal yetmezliğin diyaliz ile kontrolu ve hematokritin sabit düzeyde tutulması, trombosit fonksiyonlarını korumak için genellikle yeterlidir. Bununla birlikte hastanede takip edilen hastalardaki en büyük problem, özellikle akut renal yetmezlik zemininde gelişen üremik kanarnalardır. Üremik trombosit işlev bozukluklarının kısa dönemli tedavisi, kanama zamanını belirgin şekilde kısaltan DDAVP ve kriyopresipitat
ile yapılır. Uzun dönemli tedavide konjuge östrojenlerin de bir dereceye kadar faydası vardır. Hayatı tehdit edici kanaması ve böbrek yetmezliği olan hastalarda trombosit transfüzyonları faydalı olabilir, fakat transfüze trombositler de kısa sürede üremik defekte sahip olduklarından, bu tedavinin etkinliği de kısa sürelidir.
Kalıtılmış nitel trombosit defektIeri, trombosit reseptörleri ve granüllerindeki anormal durumları içerir. Nadir fakat iyi tamamlanmış iki trombosit reseptör bozukluğu Bernard-Soulier sendromu ve Glanvnann’s trombostenisidir. Bernard Soulier sendromu trombosit GPIb (primer vWF reseptörü) yüzey özelliklerinde azalma ve daha nadiren GPlb fonksiyonlarında bozulma ile ortaya çıkar. Sendrom hafif trombositopeni, artmış kanama zamanı, büyük trombositopeni, hafif-orta derecede kanama bozuklukları ile karakterizedir. Teşhis genellikle çocuklarda yapılır, fakat bazı durumlarda semptomlar erişkin çağına kadar ortaya çıkmayabilir. Berrnard Soulier sendromunun laboratuvar bulguları arasında; normal ristosetin kofaktör (Rcof) aktivitesi ve yeterli sayıda vWF düzeyleri ve fonksiyonu bulunmasına rağmen, ristosetine karşı trombosit agregasyonunun olamaması yer alır. Glanzmann’s trombositopenisi; artmış kanama zamanı ve trombosit GPIlbllIla özelliklerinin (fibrinojen ve vWF için reseptör) anormal düşük düzeyleri ya da daha nadir olarak normal özellikte fakat bozuk GPIlbllIla fonksiyonları ile karakterizedir. Hastalarda sıklıkla çocukluk çağında kanarna görülür. Glanzman’s trombositopesinde trombosit agregasyon testi ristosetin dışındaki bütün agonistlere cevapsızdır ya da bozuk cevap gösterir. Hem Bernard Soulier sendromunda hem de Glanzman’s trombositopenisinde trombosit transfüzyonları kanamayı düzeltir. Bununla birlikte sık yapılan trombosit transfüzyonlarının yüksek alloimmünizasyon riskinden dolayı bu tedavi çok nadir kullanılmaktadır. Kalıtımsal trombosit granül bozuklukları, eksik ya da hasarlı olan granülün tipine göre tanımlanır. Depo havuz hastalığı yoğun granüllerin eksikliği ya da göreceli olarak azlığı ile ilişkili hafif ya da şiddetli mukozal kanarnalarla karakterizedir. Yoğun granüllerdeki bozukluktan dolayı granül içeriklerinin salınımında ve trombositlerin aktivasyonunda hasar vardır. Bunun yanı sıra depo havuz hastalığında, agonistlerin büyük çoğunluğuna cevapda hasarlı ya da eksilç sekonder dalgalanma agregasyonu karakteristiktir. Hermansky-Pudlak Sendromu, okulakutanöz albinizm ve hafif trombositopeni ile ilişkili benzer yoğun granül eksikliğini gösterir. Hastalarda daha sık olarak cerrahi prosedürlerle ilişkili fakat spontan olarak da oluşabilecek belirgin kanarnalar vardır. Chediac-Higashi sendromu, nadir görülen, hafif kanarna, kısmi albinizm ve tekrarlayan piyojen enfeksiyonlarla karakterize granül bozukluğudur; nötrofillerde ve monositlerde büyük, düzensiz, grimavi renkli inklüzyonlar görülür. Gri trombosit sendromu, periferik örneklerde normal boyanmanın olmaması sonucu renksiz veya gri renkli trombositler ile karakterizedir; elektron mikroskop ile alfa granüler ve/veya içeriklerindeki kayıp doğrulanabilir. Gri trombosit sendromu olan hastalarda hafif kanama öyküsü vardır; epinefrin, ADP ve kollajene karşı agregasyon testleri bozulmuş cevap verir. Bütün trombosit granül bozuklukları; aspirin ve diğer antitrombosit ilaçların kesilmesi, kadınlarda menstürasyonların hormonal kontrolü ve kanama oluştuğunda trombosit transfüzyonları ile başarılı bir şekilde tedavi edilebilir.
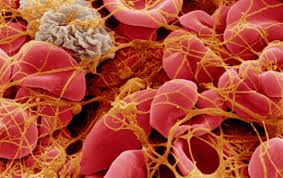
Damarsal trombosit adezyonunun fonksiyonel ligandları olan plazma proteinlerinin bozuklukları;·klinik olarak trombosit ya da vasküler bozukluklar (epistaksis, Gİ kanama vb.) ile ilişkili kanarnalara benzer tablo gösterirler. vWF endotel hücreler ve megakaryositlerde sentezlenir; plazmada hasarlı damarlara trombositlerin yönelmesini ve arkasından hasarlı bölgeye trombosit adezyonunu sağlayıcı fonksiyon görürler. vWF, farklı büyüklükte multimerik proteinleri oluşturmak için polimerize olan büyük bir moleküldür; en büyük multimerler en fazla sayıda adeziv alan içerdiklerinden küçük vWF moleküllerine göre daha fazla hemostatik etkinlik gösterirler.Anormal ya da düşük sayıda vWF düzeyi olan hastalarda hasarlı damarlarda trombosit adezyonu gecikmiştir ve bu durum uzamış kanama zamanı ile ilişkili mukozal kanamayla sonuçlanır. vWF aynı zamanda faktör Viii için taşıyıcı proteindir; vWF eksikliği ya da vWF-F VIII anormal bağlanması F VTIl’in hızlı eksikliğine yol açar, FVITI düzeyleri azalır ve PTT uzar. vWF geninde birçok farklı mutasyonlar tanımlanmıştır; bunlar fenotip olarak üç ana vWD alt grubuna ayrılır. Tip 1 vWD olan birçok hastada bütün vWF multimerlerinde hafif ya da orta derecede nicel azalma görülür. Bu durum sıklıkla heterozigot mutasyonla oluşur ve dominant kalıtım şekli gösterir. Tip i vWD; FVIII, vWF antijen ve Rcof aktivitesinde eşit derecede azalmayla karakterlidir; Rcof hasta plazmasının (vWF içeren) Ristosetin varlığında normal trombositlerle aglütinasyon yeteneğini ölçer. Tip vWD olan hastalarda sıklıkla sadece cerrahi ya da denta 1 prosedürlerle ilişkili hafif ya da orta derecede kanama vardır. Klasik olarak tip i vWD hastaları vWF’den zengin kriyopresipitat ile tedavi edilir. Bununla birlikte kriyopresipitat viralolarak inaktive ediIemediğinden günümüzde alternatifler kullanılmaktadır. DDAVP endotel hücreleri depo vWF salınımı açısından uyarır ve plazma vWF antijenlerinde, Rcof ve FVIII düzeylerinde yükselme yaratır. 0.3 mcg/kg subkutan uygulanan DDAVP, sıklıkla tip i vWD’de mükemmel sonuçlar verir. Bununla birlikte DDAVP tekrarlayan dozları sonrası endoteliyal hücrelerin yeni vWF sentezi için zaman gerektiğinden, DDAVP’ye taşiflaksi ortaya çıkabilir. Ek olarak; daha ciddi hemostatik değişiklikler gösteren tip 1 vWD olan hastalarda bazen vWF konsantreleri kullanılmalıdır.
Viral inaktive edilmiş orta derecede pürifiye FVIII ürünleri (rekombinant ya da monoklonal antikor püriyeleri değil) büyük oranlarda vWF (humate-P gibi) içerirler ve DDAVP sonrası tercih edilen tedavi şeklidir. Tip 1 vWD hastalarında gebelik sırasında kanama çok çok nadirdir. vWF düzeyleri gebelikte belirgin olarak yükseldiğinden vWF antijen ve Rcof düzeyleri sıklıkla ikinci ya da üçüncü trimestırda normaldir bu dönem için kanama riskini ortadan kaldırır. Tip 1 vWD’li gebe kadınların çoğunda doğumda kanamaya ait komplikasyon olmaz ve bunlar gebelik boyunca ya da erken post-partun dönemde tedaviye ihtiyaç göstermez. Tip II vWD çok çeşitli vWF molekülünde nitel defektler oluşturan heterozigot mutasyonlarla karakterizedir; en sık görülen tip TI bozuklukları büyük vWF multimerlerinin göreceli eksikliği ile karakterizedir. Tip IlA hastalığında elektroforezle yüksek ve orta moleküler ağırlıktaki vWF multimerleri eksiktir ve trombosit ilişkili fonksiyonlar orta derecede azalmıştır; tip Ila vWD’li hastalarda, vWF antijeni ile kıyaslandığında orantısız biçimde düşük Rcof aktivitesi görülür. Tip Ila vWD hastaları, vWF konsantrelerine ve daha az sıklıkla DDAVP’ye cevap verir. Tip IIB vWD’de anormal vWF molekülü trombositler için artmış afinite gösterir, bu durum yüksek moleküler ağırlıklı multimerlerin dolaşımdan kaybına yol açarak sıklıkla trombositopeni oluşturur. Tip llE vWD’deki trombosit agregometrisi, düşük doz ristosetin bağımlı trombosit aglütinasyonunda anormal yükselme gösterir; laboratuvarda hasta vWF’ine normal trombositlerin eklenmesi benzer şekilde ristosetin bağımlı trombosit agregasyonunu artırır ve anormal vWF’İ doğrular. DDAVP, tip IIB vWD’li hastalarda anormal vWF salınımını artırır, bu açıdan kullanımı kontroendikedir; bunun yerine vWF konsantratları kullanılabilir.
Tip IlM vWD laboratuar bulgularında tip TIA ile benzer şekilde azalmış trombosit bağımlı fonksiyon gösterir, fakat elektroforezde yüksek moleküler ağırlıklı multimerler vardır. Bu nadir vWF tipindeki bozukluk, sıklıkla vWF 'in trombosit ligandı Gplbu’ye bağlanmasının azaldığı bir mutasyondur. Tip TIM vWD’li bazı hastalar DDAVP’ye cevap verirler fakat çoğu vWF konsantresine ihtiyaç duyar. Tip lIN vWD’de anormal vWF molekülü faktör VIlFe düşük bağlanma afinitesi gösterir, karakteristik olarak faktör VIII yıkılımı artar ve hemofili A’dakine benzer bir fenotip oluşur. Gerçek hemofili A’dan farklı olarak düşük faktör VIII düzeyleri yüksek püriye faktör vın infüzyonlarına cevap vermez fakat vWF konsantratları ile iyileşme gösterir. Rcof ve vWF antijen düzeyleri, tip TIN vWD’de normaldir. Çünkü faktör VILI bağlanma bölgesindeki mutasyon vWF fonksiyon ve yaşam süresini etkilemez. Fenotip olara tip lIN kolaylıkla hemofili A ile karışabilir; vWF’in faktör VIII’e bağlanmasını saptayan testler referans laboratuvarlarında yapılabilir. Tip III vWD’Ii nadir hastalarda, sıklıkla iki anormal vWF allelinin (heterozigot bileşik) kalıtımı sonucunda, tam vWF eksikliği vardır. Tip III VWD’li hastalarda, hem Rcof hemde vWF antijen düzeyleri çok düşüktür yada hiç yoktur; Factor VIII düzeyleri %3-%10 arasındadır ve sıklıkla hemofiliyi taklit eden şiddetli kanamalar vardır. Tip III vWD DDAVP’ye cevap vermez ve kanama için vWF konsantreleri gerekir. vWD edinilmiş bir defekt olarak da karşımıza çıkabilir, genellikle şiddetlidir, bir hastada kanama hikayesi olmadan büyük vWF multimerlerinin eksikliği ile birlikte olan tip 2A benzeri bir rahatsızlıktır. Edinilmiş vWD; büyük vWF multimerlerinin anormal yılaım ile oluşur ve sıklıkla monoklonal gammopatiler, lenfoproliferatif bozukluklar, miyeloma ve diğer malign ve trombositozis ile karakterli miyoleproliferatif hastalıklarla ilişkilidir. Bu vakalarda, edinilmiş vWD, IVIG ve altta yatan hastalığın tedavisi ile başarılı bir şekilde tedavi edilebilir. Edinilmiş vWD ile sonuçlanan anormal vWF multimer yıkımının bir diğer nedeni; başarılı cerrahi tamir ile düzeltilebilecek ciddi aort stenozudur.
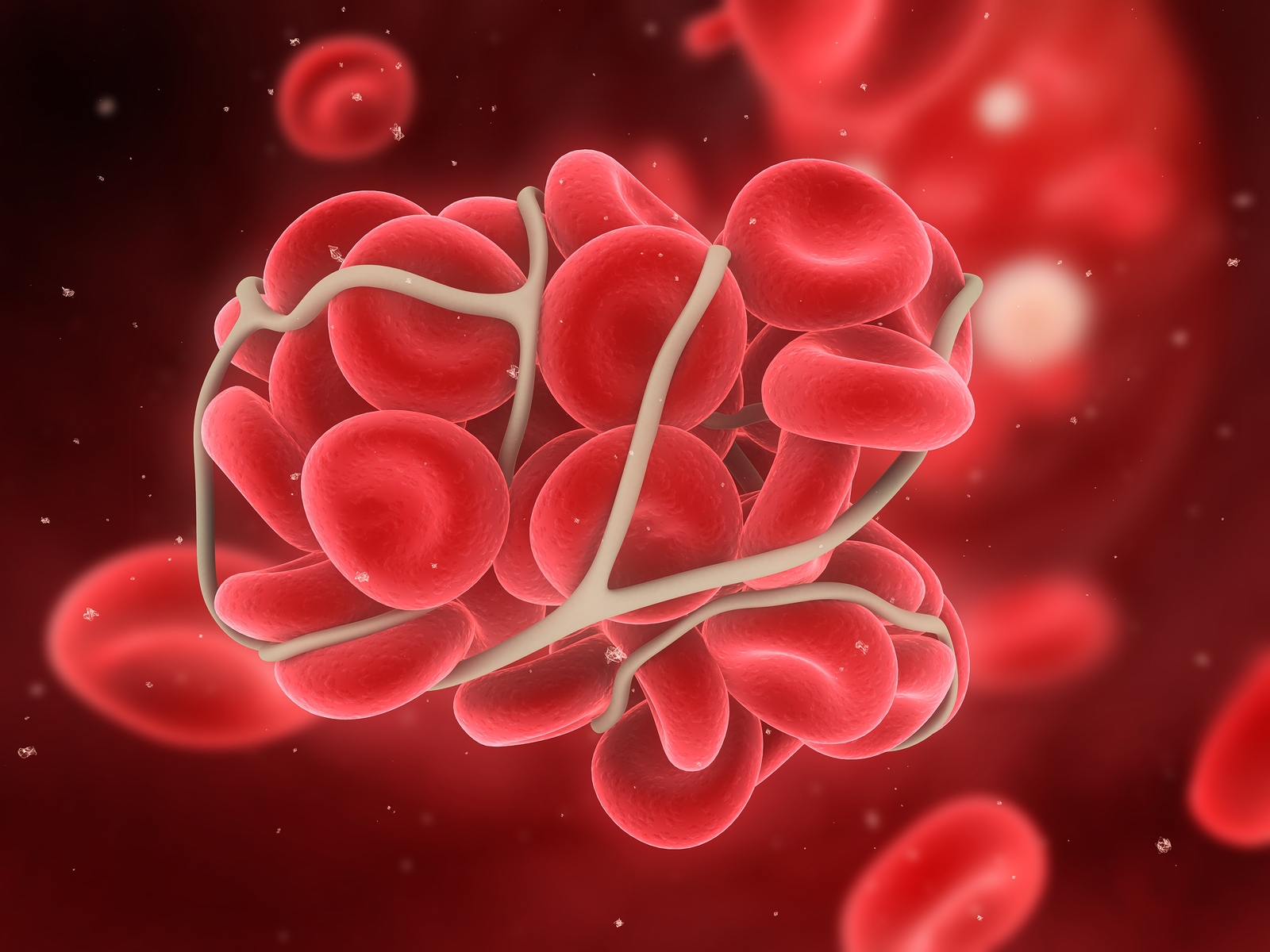
Fibrinojen, vasküler hasar bölgelerinde trombosit-trombosit matriksindeki trombosit reseptör GPIIblIIIa için köprü ligamenti gibi fonksiyon görür. Fibrinojen aynı zamanda fibrin pıhtısını oluşturacak koagülasyon mekanizmasının son basamağıdır. Her ne kadar nadir konjenital hipofibrogenemias ve afibrinogenemias da görülse de; fibrinojen düzeylerinde düşüklük, sıklıkla DIC gibi tüketim bozukluklarında görülür. Disjibrinojenemi, anormal fibrinojen proteini olarak tanımlanır. Disfibrinojenemi olan hastalarsıklıkla azalmış adesiv fonksiyonlardan dolayı kanarlar fakat bazı hastalarda hiperkoagulable durum mevcuttur. Disfibrinojenemi genellikle kalıtılır fakat daha sıklıkla karaciğer hastalıklarına bağlı ortaya çıkar. Hem PT hemde PTT, fibrinojen niteliği ve fonksiyonundaki anormalliğe bağlı olarak uzamıştır. Heparin ve fibrin yıkım ürünleri gibi inhibitörler trombin zamanını uzatsa da, bu durum düşük fibrinojen düzeyleri yada anormal molekül yapısı için daha spesifik bir durumdur. İkilenme zamanı, heparine duyarsızdır, örneğe heparin kontaminasyonundan kaynaklanan artmış trombin zamanı olasılığını ortadan kaldırmak için kullanılabilir. Hem hipofibrinojenemi hem de disfibrinojenemi, fibrinojen için zenginleştirilmiş kan ürünü olan kriyopresipitat ile tedavi edilebilir.
Normal trombosit fonksiyonları ile primer hemostaz vasküler lezyonların tıkanmasını ve mukozal bütünlüğün sürdürülmesini sağlar. Bununla birlikte, koagülasyon faktörlerinde bozukluk varsa, başlangıçtaki trombosit tıkacı normal sekonder hemostaz ile sıkılaştırılamaz ve pıhtı bozularak kanama başlar. Bu kanama trombosit tipi kanamadan farklıdır; koagülasyon eksiklikleri derin dokularda ve eklemlerde kanama yaratır ve hafif eksiklik durumlarında da cerrahi sonrası gecikmiş tipte kanamalar oluşabilir. Her ne kadar hafif derecede eksikliği olan hastalar normal koagulasyon taramaları ile kanama sergileseler de; belirgin faktör eksikliği olan hastaların çoğu tarama laboratuvar testlerinde anormal sonuçlar gösterir Faktör VIII (lıemofili-A) ve faktör-IX (hemofili-B)'un X-bağımlı eksiklikleri, vWD sonrası en sık görülen faktör eksiklikleridir. Hemofili-A, hemofili-B’ye göre altı kat daha sıktır. Şiddetli hemofili-A bulunan hastaların yaklaşık %50 ya da daha fazlası; büyük oranda gen inversiyonu sonucu, gen tam aktivite kaybı sonucunda ortaya çıkar. Diğer mutasyonlar oldukça hafif hastalıklarla sonuçlanma eğilimindedir. Hemofili-B olan hastaların çoğunda, aktivite kaybı sonucu anormal faktör-IX fonksiyonlarıyla sonuçlanan mutasyonlar vardır. Antijenik ve fonksiyonel testlerin sonuçlarının değerlendirilmesi; eksikliğin protein kaybından mı yoksa proteinlerin normal fonksiyonlarının kaybından mı kaynaklandığını ortaya çıkarabilir. Hem Hemofili-A hem de hemofili-B faktör düzeylerine göre kategorize edilir: şiddetli eksiklik faktör VIII ya da IX bulunmaması «%1) ile karakterizedir,oysaki hafif yada orta derecede hemofilide faktör düzeyi %1 ila %5 arasında ve bazen de göreceli olarak %5 üzerindedir. Şiddetli Hemofili-A ve Hemofili-B, çocukluk döneminde kasIara, eklemlere ve yumuşak dokulara kanama gelişmesiyle ortaya çıkar. X-bağımlı rahatsızlıklar olduğundan primer olarak erkek hastalarda görülür; etkilenen erkek hastanın annesi taşıyıcıdır ve dayılarının %50’si aynı hastalığa sahiptir. Bununla birlikte hemofili vakalarının %25-30’u yeni mutasyonlardır ve belirgin aile öyküsü içermezler. Bu durumların çoğunda, yoğun Xinaktivasyon bozukluğu olan faktör düzeyleri <%30’un altındaki bayan taşıyıcılarda hafif kanama bozuklukları olabilir. Şiddetli hemofilide kanama sıklıkla spontandır, herhangi bir cerrahi girişim ya da hafif travma sonrasında bile görülebilir. Hemofilide kanama sıklıkla eklemlerde ve retroperitondadır; hematüri ve mukozal ve intrakranial kanama da olabilir. Orta derecede hemofilisi olan hastalarda daha az spontan kanama görülmekle birlikte cerrahi yada travmaya bağlı hemorajik komplikasyon riski belirgindir. Hafif hemofilisi olan hastalar, erişkin çağına kadar saptanamayabilir ve sadece major cerrahi girişimler sonrası kanama gelişebilir. Hemofili komplikasyonları, eklem ve kasıara olan; şiddetli deformitelere, artrite, kas atrofilerine ve kontraktürlere yol açan, kronik kanamalardan kaynaklanır; bu komplikasyonlar sıklıkla eklem replasmanı gibi yoğun ortopedik girişimler ve fizik tedavi gerektirebilir. Ek olarak; hemofili olan ve viral inaktivasyonun başlamasından önce faktör konsantreleri uygulanış olan hastalarda, transfüzyonla geçen enfeksiyonlarla; özellikle HIV, hepatit B ve C; ilişkili komplikasyonlar olabilir.Mevcut tedavide viral inaktive ya da rekombinant faktör konsantreleri kullanılmaktadır. Hızlı faktör replasmanı etkili tedavinin anahtarıdır. Şiddetli hemofilisi olan hastalar, sıklıkla düzenli bir şekilde düşük dozlarda profilaktik faktörü kendi kendilerine infüze ederler (25-40 U/kg dozda haftada üç defa) ve ilaç dozlarını yada infüzyon sıklıklarını İnternal kanama, travma yada dental prosedür durumlarında dozlarını arttırırlar. Hafif hemofili A bulunan hastalar küçük cerrahi girişimler için faktör infüzyonlarına gerek duymayabilir; bu hastalar sıklıkla 0,3 mcg/kg. DDAVP infüzyonu ile birlikte ya da tek başına her 4-6 saatte bir uygulanan f-aminokaproik asit ile tedavi edilirler. Bunun yanı sıra, hemofili hastalarının çoğu, profilaktik olmasa da cerrahi veya travma sırasında, faktör infüzyonlarına gerek duyar. Her 8-12 saatte Faktör-VIII ürünleri infüze edilir, ve 1 U/kg faktör VIII konsantresi plazma faktör VIII aktivitesini %2 arttırır; böylelikle şiddetli hemofili gösteren bir hastada teorik olarak 50 U/kg faktör VIII, %100 faktör VIII aktivitesi ortaya çıkarır. Faktör IX daha uzun bir yarılanma ömrüne sahiptir ve her 18-24 saatte bir infüze edilir; faktör IX aktivitesinde %2’lik bir artış sağlamak için 2 U/kg faktör IX’a ihtiyaç vardır (%100 aktivite için 100 U/kg gerekir). Hemofilili hastalardaki birçok cerrahi girişim; hem intraoperatif periodda hem de erken postoperatif periodda, yara hematom oluşumunu önlemek ve normal faktör değerlerini (>%80) yakalayabilmek için yoğun faktör terapisi gerektirir. Faktör dozları olayın şiddetine, hastanın önceki faktör infüzyonlarına cevabına ve faktörlere karşı inhibitör gelişip gelişmemesine bağlı olarak ayarlanır.
Faktör V, VII, X ve XI’e bağlı koagülasyon faktör eksikliklerinin yol açtığı kalıtımsal kanama bozukluklarına Hemofili A ve B’den daha az sıklıkla karşılaşılır. Faktör V eksikliği olan hastalarda genellikle hem plazma faktör V düzeyi hem de trombositfaktör V düzeyi eksiktir ve hemofili hastalarındakine benzer eklem ve kas kanarnaları gösterir. Plazma faktör V eksikliği olan bazı hastalar, cerrahi ya da travmaya bağlı stresle karşılaşmadıkları sürece asemptomatiktirler ve bu hastalarda trombosit faktör V düzeylerinin normalolduğu düşünülmektedir. Faktör V eksikliği, taze donmuş plazma ya da trombositler ile tedavi edilebilir; trombositler geneııikle, uzun dönem plazma tedavisi sonrası faktör V’e inhibitör gelişmiş olan hastalarda faydalıdır. Nadiren hastalar, faktör V ve faktör VIII eksikliği kombinasyonu gibi, faktör eksikliklerini arka arkaya kalıtabilirler. Faktör XL eksikliği (Hemofili e) gösteren hastalarda, Hemofili A ya da B’ye göre genellikle hafif kanama bozukluğu görülür (faktör XI düzeyleri <%5 olsa bile) ve bu hasta grubu plazma infüzyonlarıyla tedavi edilir. Oysaki faktör X eksikliği genellikle daha ciddidir ve aynı zamanda plazma ile tedavi edilebilir. Faktör X eksikliği Ashkenazi Yahudileri arasında artış sıklıkta görülen otozomal resesif bir bozukluktur; hemofili C sıklıkla ileri yaştaki erişkinlerde gelişir ve prostat cerrahisi sonrası gelişen artmış fibrinolizle ilişkili klinik bulgular bu duruma bir örnektir. Edinilmiş faktör X eksikliği amiloidoz ile ilişkili ortaya çıkabilir; dolaşımdaki anormal hafif zincirlerin adsorbe olduğu, faktör X yıkımının gerçekleştiği ve düşük seviyelerde nadir kanarnaların geliştiği bir durumdur. Nadir görülen faktör- VII eksikliği olan hastalarda, %10 altındaki düzeyler protrombin kompleks konsantratları ya da rekombinant faktör VIIa ile tedavi edilebilir. Purifiye ya da rekombinant faktörlerin gelişimi önemlidir çünkü taze donmuş plazma ile faktör düzeylerinin yerine konması zordur. Taze donmuş plazmadaki faktör kon-santrasyonları değişkendir ve in vivo konsantrasyonlara benzerdir; ek olarak faktör düzeyini %O’dan %30’a çıkarmak için dört ya da daha fazla ünitede taze donmuş plazma gerekir. Bu yüksek sıvı yükü; kalp hastalığı, karaciğer yetmezliği ya da renal yetmezliği olan hastalarda problem oluşturur.
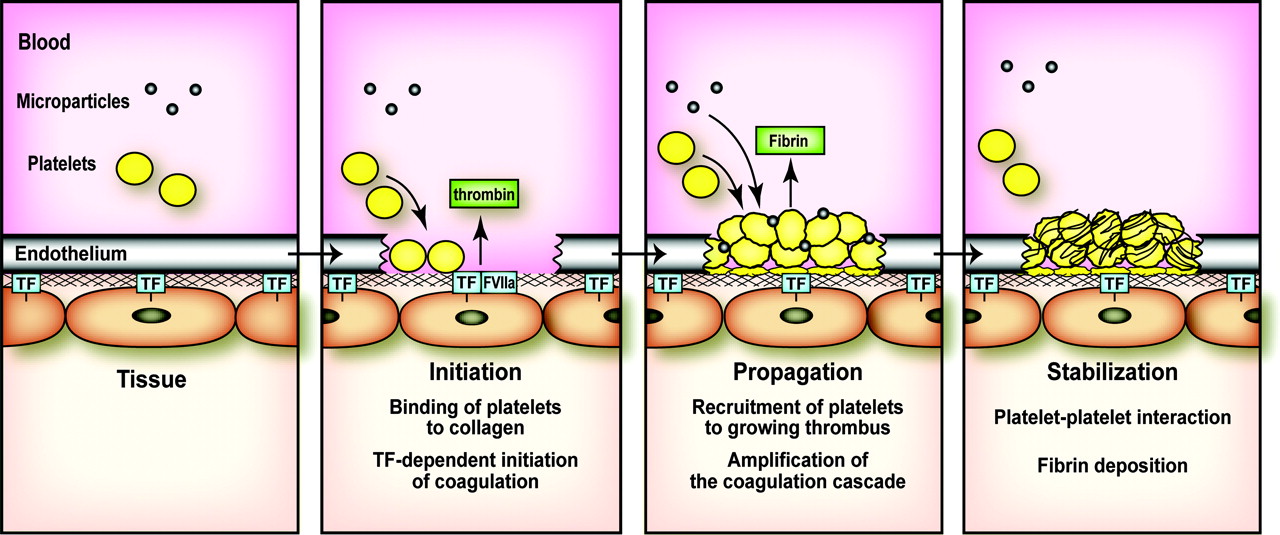
Faktör inhibitörleri hemofili-A’lı hastalann yaklaşık %25’inde transfüze faktör- VIII’e karşı otoantikodar gelişir. Antikor fonksiyonelolarak in vivo etkilidir ve in vitro olarak Bethesda unitesi (BU) ile ölçülür; BU, faktör aktivitesini %50 oranında nötralize eden inhibitör ünitedir. Yüksek düzey inhibitörler (>10BU) infuze faktör konsantrasyonlarının kanama epizotlanndaki etkinliklerini azaltarak, aktivitesini nötralize ederler. Bu açıdan kanamada; faktör VIII inhibitör bypass activitesi (FEIBA) veya rekombinant faktör VIIa gibi farklı tedavi seçenekleri kullanmak gerekir. Uzun dönem tedavide; IVIG kombinasyonu, immunsupresif tedavi, plazmaferez ve yüksek doz infuzyon konsantreleri kullanılarak immun toleransın indüksiyonu ile inhibitör supresyonu gerçekleştirilir. Hemofili-B hastalarında inhibitör insidansı daha az düzeydedir (%2-%6), fakat bu hastalar da antikorların uzun dönem supresyonu için yüksek doz FEIBA ya da rekombinant VlIa ile
benzer şekilde ve benzer stratejilerle tedavi edilir. Faktör- VIII’e karşı edinilmiş inhibitörler (diğer koagülasyon faktörlerinden daha az olarak) ara sıra, daha önceden kanama öyküsü olmayan hastalarda (sıklıkla yaşlılarda) ortaya çıkabilir. Edinilmiş faktör-VIII inhibitör titreleri, bazen oldukça yüksek düzeylerde olabilir ve sıklıkla lenfoproliferatif hastalıklar gibi malign hastalıklarla ilişkilidir. Faktör-VIII’e karşı edinilmiş inhibitör bulunan hastalar, benzer şekilde Faktör VIIa veya FEIBA ile tedavi edilirler. Yoğun immunsupresif tedavi (siklofosfamide ve prednisone ile) başarılı tedavinin başlıca dayanağıdır ve inhibitörleri yok etmek için mümkün olan en kısa zamanda uygulanmalıdır.
Hastanede yatarak ya da ayaktan takip edilen ağır hastalığı bulunan kişilerde, edinilmiş koagülasyon faktör eksikliklerinden kaynaklanan kanamalar olabilir. Faktör eksikliği nedenleri arasında en önde geleni K-vitamin eksikliğidir. K-vitamin eksikliği aşağıdaki faktörlerden herhangi birisine bağlı olabilir: (I) K-vitamini emiliminde azalmaya yol açan, enterohepatik dolaşımla ilişkili safra yolu hastalığı; (2) ilaçlar; özellikle barsaklan sterilize eden ve K-vitamini bakteriyel kaynaklarını azaltan antibiyotikler veya K-vitamin emilimini doğrudan bloke eden diğer ilaçlar (kolesteramin) (bu kategori içerisine, yağda eriyen vitaminin intrahepatik metabolizmasıyla etkileşim gösteren sefalosporinler de dahildir.); (3) malabsorpsiyon bozuklukları (sprue), kronik hastalıklar ya da kötü oral alımı olan akut hastalıklar ile ilişkili kötü beslenme durumları. Daha önceden bahsedildiği şekilde, protein C ve S gibi, faktör II, VU, IX ve X, K-vitamini bağımlı prokoagülan faktörlerdir. Warfarin; bu faktörlerin K-vitamini bağımlı y-karboksilasyonunu bloke ederek fonksiyonel faktör- VII düzeylerinde akut azalmaya yol açar; faktör-VII in vivo olarak K-vitamin bağımlı faktörler arasında en kısa yarılanma ömrü ne sahip (6 saat) faktördür. Oral veya parenteral olarak K-vitamininin yerine konulması (3 gün boyunca 1-10 mg./gün) normal karaciğer fonksiyonları dahilinde koagülasyon
faktör sentez bozukluğunu düzeltir.
Hafif ya da orta şiddette karaciğer bozukluğu olan hastalarda PT süresi uzarken, sıklıkla PTT değerleri normaldir. Şiddetli karaciğer hastalıkları ise hem PT hem de PTT değerlerini yükseltir. K-vitamini eksikliği ya da varfarin alanlardan farklı olarak; karaciğer bozukluğu olan hastalarda, sadece K vitamini bağımlı faktörler değil, istisnai olarak faktör-VIII dışındaki hemen hemen bütün faktörlerin düzeyleri düşüktür. Hemotilili hastalarda Karaciğer transplantasyonu her ne kadar faktör-VIII düzeylerini arttırsa da, faktör VIII düzeyleri genellikle karaciğer hastalığı ile birlikte yükselir, bu bulgu karaciğer dışında başka kaynaklardan VIII üretimi ile ilişkilidir. Karaciğer rahatsızlığı olan bir hastada faktör-VIII düzeyleri azalıyorsa, olaya DIC tablosunun eklenebileceği akılda tutulmalıdır. Bu açıdan uzamış PT sebebi değerlendirilirken, faktör- VU ve faktör V gibi diğer faktörlerin düzeylerinin tespiti de faydalıdır. K-vitamini eksikliklerinde, faktör-VII düzeyi düşük, faktör -V düzeyi normaldir; zıt olarak generalize karaciğer hastalıklarında hem faktör- V hem de faktör- VII düzeyleri düşük olabilir. PT karaciğer fonksiyonlarının duyarlı bir göstergesidir ve hafif karaciğer bozukluğu olan hastalarda bile yüksek bulunabilir; bu yükselme albumin ya da prealbumin düzeylerinde belirgin bir düşme oluşturur ve sıklıkla transaminaz değişiklikleriyle beraberdir. Hafif ya da orta derecede karaciğer bozukluğu olan hastalarda, PT uzamıştır fakat PIT genellikle normal sınırlardadır. Şiddetli karaciğer bozukluğu geliştiğinde, PT düzeyi daha da belirgin yükselme gösterir ve PTT düzeyi de beraberinde anormal değerlerdedir. Karaciğer hastalıklannda, faktör sentezindeki azalma dışındaki, kanama sebepleri: (1) fibrin yıkım ürünlerinin atımının azalması ve/veya ilişkili DIC, (2) trombosit fonksiyonlarının inhibisyonu ve (3) doku plazminojen aktivatör düzeylerinde artma olarak sıralanabilir. Taze donmuş plazma ile koagulasyon faktörlerinin yerine konulması mevcut tedavi seçeneğidir, fakat rekombinant VIIa, karaciğer bozukluklarına bağlı kanamaların tedavi ve profilaksisinde ümit verici sonuçlar vermektedir.
Bazen, kanama bozukluğu olan hastalar laboratuvar tarama testlerinde (PT, PTI, trombosit sayısı vb.) herhangi bir bozukluk göstermezler. Önceden belirtildiği gibi, bu bozukluklar vasküler purpuralan içerse de, diğer bazı kanama bozuklukları da bu grup içerisinde yer alır. Hafif vWD’e bağlı kanaması olan hastalarda PTI normalolabilir; fakat çalışmalarda genellikle faktör VIII, vWF antijen yavda vWF Rcof düzeylerinde hafif düşüklük görülmektedir; hafif tip 2A vWD olan hastalarda çok merkezli analizler de anormal olabilir. Benzer şekilde, hafif faktör eksiklikleri (faktör II, V, VII, VIII, IX, yada XI) PT veya PTT düzeylerini uzatmayabilir, fakat spesifik faktör testleri normalden daha düşük düzeyler göstermektedir. Cerrahi veya travma sonrası geç başlangıçlı hafif kanama; faktör XIII eksikliği yada disfibrinojenemiden kaynaklanan pıhtı instabilitesi olan hastalarda ortaya Çıkabilir; neonatallerde faktör XTII eksikliği geç dönem umblikal kanamalarla gelişebilir. Faktör XIII eksikliği, idrarda artmış pıhtı çözünürlüğü ile sonuçlanır; şayet pıhtı 8 mol/L idrarda çözünürse, XIII düzeyi için ELİSA testi uygulanabilir. Faktör-XIII eksikliği, taze donmuş plazma ile tedavi edilir. Düşük fibrinojen düzeyleri ya da anormal fibrinojen fonksiyonları, hem trombin zamanını hem de reptilaz zamanını uzatır. Trombin zamanı aynı zamanda heparin ile de uzar fakat reptilaz zamanı heparin duyarlı değildir ve bu açıdan heparin kontamınasyon varlığının dışlanmasına; ya da test fibrinojen düzeyleri ve fonksiyonlarının heparin varlığında gerçekleştirilmesine olanak sağlar. Son olarak; normal trombosit sayısı ve pıhtılaşma zamanı olan kanamalı hastalar, trombosit nitel fonksiyon testleri ile değerlendirilebilir; trombosit reseptör ya da granüllerindeki kalıtımsal eksikliklerde ve üremi ya da ilaçlar ile oluşan trombosit anormalitelerinde, anormal trombosit agregasyonu veya uzamış kapanma zamanı saptanması ile tanı konulabilir.