
Mitokondriyal bozukluklar yaşlanma kadar, bazı metabolik ve nörodejeneratif hastalıklarda da rol oynamaktadır. Hemen hemen bütün organ sistemleri oksidatif metabolizmayı kullandığından mitokondriyal hastalıklar çok yönlü klinik belirtilere sahiptir. Klinik özellikler merkezi ve periferik sinir sistemi, göz, kas, böbrek ve endokrin organlar gibi yüksek enerji gerektiren dokularda sık sık ortaya çıkar. Mitokondri hastalıklarının.başlama yaşı, biçimi ve klinik seyri nukleus genomununkinden belirgin olarak farklı olan mitokondri DNA’sının (mtDNA) replikasyon mekanizması nedeniyle değişiktir. Birçok mitokondri hastalığında maternal kalıtım biçimi karakteristiktir, çünkü mtDNA oositlerle aktarılır. İlk mutasyonun tanımlandığı 1988’den bu yana, yüzlerce farklı mtDNA mutasyonu tanımlanmıştır.
MİTOKONDRİ DNA’SININ YAPI VE FONKSİYONU
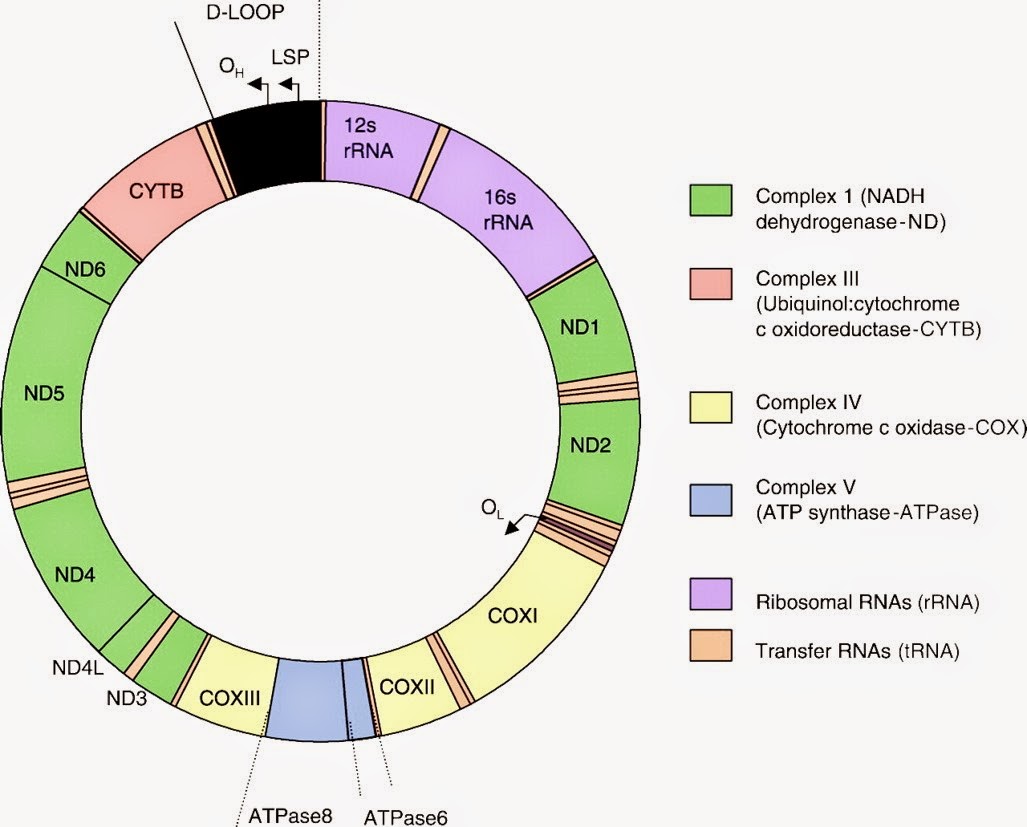
Sayısı dokunun enerji gereksinimi ve fonksiyonuna bağlı olmak üzere, çoğu hücreler yüzlerce mitokondriye sahiptir. Mitokondriler kendi ekstrakromozomal DNA’sını taşıyan tek hücre organelidir. İnsan mtDNA’sı küçük (16.569bç), çift iplikli ve halkasal bir molekül olup 4 farklı biyokimyasal oksidatif fosforilasyon kompleksinin 13 protein alt birimini kodlar. mtDNA aynca bu proteinlerin intramitokondriyal translasyonu için gereken 24 yapısal RNA’yı (2 ri- bozomal RNA ve 22 transfer RNA) da kotlar. Kotlanmayan D (Displacement =yerdeğiştirme) ilmeği transkripsiyon ve replikas- yonu kontrol eden regülatör bölgedir, mt DNA mutasyonlan her tipteki mitokondri geninde bulunur.
Mitokondriler muhtemelen bağımsız bir organizmadan gelerek hücrenin içine simbiyotik olarak dahil olmuştur. Sonuç olarak mitokondriler, DNA’smın replikasyon, transkripsiyon ve translasyo- nunu nükleer DNA’dan bağımsız olarak yapar. Bununla beraber, hücresel ve mitokondriyal fonksiyon birbirine bağımlıdır. Nükleer DNA tarafından kotlanan proteinler oksidatif fosforilasyonla da ilgilidir ve mitokondrinin yapı ve fonksiyonu için gerekli sayısız makromoleküler bileşikler (örn, mtDNA replikasyonu, transkripsiyonu ve translasyonu) mitokondriye sitoplazmadan taşınır.
Oksidatif fosforilasyon ve enerji gerektiren hücresel olaylar için adenozin trifosfat (ATP) üretimi mitokondriyonların başlıca görevidir. Mitokondri fonksiyonundaki değişiklikler üç ana mekanizmayla hastalık patogenezine sebep olurlar: (1) mutasyonlar oksidatif fosforilasyonu bozduğunda ATP oluşumundaki azalma; (2) DNA, protein ve yağlara hasar veren OH-serbest radikallari ve H2O2 gibi reaktif oksijen türlerinin açığa çıkarılması; ve (3) mito- kondriyonlar, kaspaslar (caspases), sitokrom-c ve apopitoz başlatı- cı faktör gibi hücre ölümünü uyaran faktörler salgıladığında apopitoz yolunun çalıştırılması,
mtDNA’mn bazı özgün özellikleri onu mutasyonlara karşı hassas hale getirir ve hastalıklardaki rolüne katkı yapar. Örneğin, mtDNA’da intronlar yoktur (bu durumda rasgele bir mutasyon, çoğunlukla kotlanan DNA dizilerini vurur) veya koruyucu histonlar yoktur ve mükemmel olmayan bir DNA tamir sistemine sahiptir ve oksidatif fosforilasyonla ortaya çıkan serbest oksijen radikallerine maruz kalır. mtDNA’mn nükleer DNA’ya göre 10 defa daha hızlı mutasyona uğradığı tahmin edilmektedir.
Önemli bir husus da mtDNA’mn kesinlikle maternal olarak kalıtılması ve rekombinasyona girmemesi ve sonuçta mtDNA mutasyonların anne soyu boyunca ardışık olarak birikmesidir. Bu özellikler mtDNA dizisindeki varyasyonları, adli tıpçı ve evrimsel biyologlar açısından bulunmaz birer araç durumuna getirir. Mendel genetiği karşıtlığına rağmen, miıokondfiyal genetiği populasyon genetiği ilkeleri yönetir. Yeni bir mtDNA mutasyonu olduğunda, hücreler başlangıçta normal ve mutant DNA dizilerini birlikte bulundurur. Bu durum heteroplazmi olarak bilinir bu da aksi halde ölümcül ve hastalığa sebep olacak olan mutasyonlann devamını sağlar. Tamamiyle normal veya tamamen mutant mtDNA’nın birlikte bulunmaları homoplaztni olarak bilinir. Hücre bölünmesi esnasında, replikatif dağılım (segregasyon) denen olayla mitokondriyonlar, yavm hücrelere eşit olmayacak şekilde paylaştırılır; sonuç olarak mutant ve normal mtDNA moleküllerinin oranı değişebilir. Seleksiyon baskısı moleküler, hücresel ve organizma düzeyinde etkilidir. Zararlı fenotipik ekspresyon olması için gerekli mutant mtDNA’nın kritik oranı eşik etki olarak bilinir. Bu oran, oksidatif arz ve talepteki hassas dengeye bağlı olarak kişiler arasında, organ sistemleri ve belli bir doku içinde farklılık gösterir. Hücre bölünmesi esnasında mitokondriyonlann yavm hücrelere eşit olmayan bir biçimde aktarılmasının eşlik ettiği, mtDNA ayrılmasındaki bu özellikler mitokondriyal hastalıklardaki fenotip farklılığının temelini oluşturur.
MİTOKONDRİYAL DNA MUTASYONLARI
Şiddetli, ölümcül derecede oksidatif fosforilasyon azalmasına sebep olan mtDNA mutasyonlan büyük yapısal bozukluklar veya yapısal RNA’lardaki nokta mutasyonlan gibi sadece heteroplazmik ise yaşayabilir. Aksine, protein kotlayan genlerdeki hafif, missens (yanlış anlamlı) mtDNA mutasyonlannın çoğu homoplazmiktir. mtDNA mutasyonlan mtDNA genlerinin her birinde bulunmuştur, fakat tRNA mutasyonlan şiddetli multisistemik mitokondriyal ensefalomiyopati fenotiplerinde daha fazladır; protein kodlayan gen mutasyonlan kalıtsal Leber optik nöropatisinde (LHON) daha fazladır. 12S ribozomal RNA genindeki bir nokta mutasyonu, kendiliğinden olan ve aminoglikoza bağlı olarak gelişen duyu siniri sağırlığının her ikisi ile de ilişkilidir.
MtDNA ve klinik fenotip arasında, tanımlanmış bir sebep sonuç ilişkisi kurmak çeşitli sebeplerden dolayı çok zordur: (1) mtDNA oldukça polimorfiktir, (2) faiklı mutasyonlar aynı fenotipi ya da aynı mutasyonlar farklı fenotipleri ortaya çıkanrlar ve (3) epigenetik faktörler klinik belirtileri etkileyebilir.
KLASİK MİTOKONDRİYAL ENSEFALOMİYOPATİ FENOTİPLERİ
Anormal mitokondri morfolojisi, biyokimyası veya maternal kalıtım biçiminden dolayı çoğu hastalıklar geçici olarak mitokondriyal bozukluklar olarak sınıflandırmışlardır. Hastalıkla ilgili mtDNA mutasyonlan şimdi tanı kriteri açısından önemlidir. Her klasik mitokondriyal ensefalomiyopati fenotipinin belirgin klinik özellikleri olmasına karşın bunların her biri çoğu klinik ve laboratu- var özellikleri de paylaşırlar.
Mitokondriyal miyopati eksersize karşı dayanıksızlılık kendini gösteren kalıcı proksimal zayıflıkla karakterizedir. Yorgunluk ve hafif kuvvetsizlik göze çarpan klinik özelliklerdir, fakat mitokondriyal etiyoloji diğer nörolojik, somatik veya laboratuvar bulgularıyla değerlendirilir. Frank rabdomiyolizi seyrektir. Elektromiyografi irritasyona neden olmayan bir miyopati olduğunu belirtir ve serum keratin kinaz çoğunlukla normal veya hafif yükselmiştir. İskelet kası biyopsisi anormal mitokondri bölünmesini gösterir ve ”pürtüklü kırmızı fibriller" oksidatif fosforilasyondaki şiddetli biyokimyasal bozuklukların histolojik olarak belirleyicisidir. Mitokondriyal miyopatide, büyük mtDNA delesyonlan ve çeşitli mtDNA nokta mutasyonlan oluşur.
Kronik ilerleyen eksternal oftalmopleji (CPEO) Ptozis, oftalmopleji ve ekstremde miyopatisi CPEO yu karakterize eder. Diğer klinik özellikler mitokondriyal hastalıklara özgü laboratuvar ile birlikte de olabilir. Kırmızı lifli anormal iskelet kası biyopsilerine ve ultrasütrüktür değişikliklere sahiptir. CPEO ile ilişkili olarak ilave merkezi ve somatik sinir sistemi bulgularına sahip olanlar ”CPEO-plus” sendromu olarak bilinirler. Keams-Sayre sendromu CPEO ve atipik pigmenter retinopati ile karakterize ve 20 yaştan önce başlayan CPEO-puls’ın bir alt gmbudur; yardımcı özellikler yükselmiş serebrosipinal sıvı proteini, ataksiya veya kalp bloğudur,
CPEO lu hastaların çoğu, mtDNA da moleküler genetik yöntemlerle doğru olarak belirlenebilen büyük, tek delesyonlara sahiptir. Bununla birlikte, hemen hemen bütün tek mtDNA delesyonlan sporadik olarak ortaya çıktığından, DNA kaynağı olarak iskelet kası gerekir. Pigmenter retinopatinin varlığı şiddetle, bir delesyonun varlığını işaret eder. Replikasyon esnasmda rekombinasyon ve kayma olasılığı olduğu halde, DNA delesyon mekanizması bilinmemektedir. Çoğu delesyonlann birleşme noktalan, bütün delesyonların yaklaşık %25 inden sorumlu olan "sıcak nokta” denen 13 nukleotidlik tekrarlar dahil direkt tekrar dizilerine sahiptir. Yaklaşık olarak delesyonlann yansı diğer direkt tekrarlarla ilgilidir. Belirgin olarak direkt tekrara sahip değildir. Çok az hasta kısmen duplike olmuş mtDNA molekülüne sahiptir, mtDNA delesyonu veya duplikasyonu taşımayan çok sayıda hastada 3243. nukleotid pozisyonunda bir nokta mutasyonu bulunur.
Otozomal olarak geçen çoklu mitokondriyal DNA delesyonları klinik olarak değişik tip CPEO lu birkaç ailenin iskelet kaslarında çoklu mtDNA delesyonu taşıdıkları bulunmuştur. Çoklu mtDNA delesyonlannın otozomal kalıtımı mtDNA üzerinde sekonder etki yapan nükleer DNA daki primer bozukluğu işaret eder. Çoklu mtDNA delesyonları otozomal dominant veya otozomal resesif biçimde geçebilir veya somatik mutasyon olarak meydana gelebilir.
Timidin fosforilaz normal mtDNA nın regülasyon ve fonksiyonunu trans-aktif biçimde etkileyen çekirdek tarafından kotlanan birinci gendir. Timidin fosforilazdaki mutasyonlar miyonöragastrointestinal ensefalomiyopati (MNGIE) denilen otozomal reşesif bir hastalığa sebep olur. CPEO nun otozamal dominant formları çeşitli farklı nükleer lokuslarla bağlantılıdır. Doku spesifik otozomal olarak kalıtılan mtDNA nın kaybı, genomlar arası iletişim hatasının sebep olduğu kantitatif mtDNA kusurunu temsil eder.
Mitokondriyal Ensefalomiyopati, Laktik Asidoz ve İnme Benzeri Nöbetler (MELAS) Bu sendrom subakut beyin disfonk- siyonu, serebral yapısal değişiklikler, nöbetler ve diğer sık rastlanan klinik ve laboratuvar bulgulatma yol açan inme benzeri olaylarla karakterizedir. MELAS sendromunun maternal kalıtımı yakın akrabalardaki klinik özelliklerinin hafif olmasından dolayı gizlenebilir. tRNA Leu (UUR> geninin 3243. nukleoti- dindeki bir nokta mutasyonu MELAS olgularının %80’inin sebebidir. Bununla birlikte, 3243’deki mtDNA mutasyonunun klinik özellikleri pleimorfiktir; aynı zamanda delesyonsuz CPEO da miyopati, sağırlık, şeker ve distoni ile ilişkilidir.
Pürtüktü Kırmızı Fibrilli Miyoklonik Epilepsi (MERRF) Sendromu MERRF sendromu miyoklonik deşarjlar, nöbetler, serebellar ataksiler ve mitokondriyal miyopatilerden oluşmuştur. Patogenetik mutasyonlar, tRNAL7s geninin 8344. ve 8356. pozisyonundaki nukleotidlerde gösterilmiştir. Diğer mitokondriyal ensefalomiyopatilerde sık rastlanan nörolojik ve laboratuvar özellikler görülür. Anne akrabaları asemptomatik olabilir veya karakteristik olarak “at yelesi” tarzında dağılmış lipomlar ve yüksek tansiyon dahil kısmi klinik sendromlara sahip olabilir.
Nöropati, ataksi ve retinitis pigmentosa (NARP), maternal olarak kalıtılan leigh hastalığı NARP proksimal güçsüzlük, duyusal nöropati, gelişmede gecikme, ataksi, nöbetler, demans ve retinadaki pigment bozukluğu ile karakterizedir. Anne tarafından kalıtılan bu hastalık ATPaz 6 geninin 8993. pozisyonundaki iki farklı heteroplazmik missens mutasyonla ilişkilidir. Yüksek orandaki aynı mutasyonlar maternal olarak kalıtılan Leigh hastalığında da görülür. Otozamal resesif Leigh hastalığı sitokrom c oksidaz eksikliğinden olur ve buna sitokrom c oksidaz kompleksinin (kompleks IV) biyogenezi için gerekli olan nukleusta kodlanan SURF1 proteininin eksikliği neden olur. NARP, MELAS, MERRF ve diğer mitokondri hastalıklarına sebep olan mtDNA mutasyonlan kan veya kaslardan izole edilen mtDNA nın moleküler genetik analizleriyle halihazırda tetkik edilebilir,
Leber kalıtsal optik nöropati LHON tipik olarak ağrısız, subakut, merkezi skotoma ve diskromatopsi ile oluşan bilateral görüş kaybı ile kendini gösterir. Ortalama başlama yaşı 23 tür ve erkekler kadınlara göre 3-4 defa daha fazla olarak etkilenirler. LHON diğer mitokondriyal hastalık fenotiplerine klinik olarak çok az benzerlik gösterir. Bu anne kalıtım biçimi bazında ilk kez bu grup içinde sınıflandırılmıştır. Görme kaybının fizyopatolojisine hem genetik ve hemde epigenetik (tütün ve alkol) faktörler birlikte dahil oluyor gibi görünmektedir. mtDNA mutasyonlan yüksek derecede heterojenlik göstermektedir. LHON la ilgili primer mtDNA mutasyonlan çoğunlukla kompleks 1 genlerini ve 14484 (ND-6 geni)] nukleotid pozisyonlannda etkiler (Şek.67-1). Kafkas haplogrup J mtDNA sımn 13708: (ND-5 geni) nukleotid pozisyonundaki bir mutasyon da dahil diğer birçok mtDNA mutasyonlan LHON tâki sekonder patogenetik rollere sahip olabilir.
Primer LHON-ilişkili mtDNA mütasyonları arasında genotip-fenotip korelasyonlan ortaya çıkmaya başlamıştır. Örneğin, görmen iyileşimi prognozu mutasyona bağlı olarak yaklaşık 10 defa değişir. LHON la birlikte distoniye sebep olan mtDNA mutasyonlan da tanımlanmıştır.
MİTOKONDRİ HASTALIĞININ ORGAN SİSTEMİ BELİRTİLERİ
Hemen hemen bütün vücut dokulan bir dereceye kadar oksidatif metabolizmaya bağlı olduğundan, mitokondriyal hastalığı olan hastalar tıpdaki çoğu uzmanlara başvurabilirler. Öncelikle klasik mitokondri hastalıktan ile ilişkili olduğu belirtilen olan somatik belirtiler dominant olabilir veya klinik semptomun başlangıcı veya önemli komorbit özellikler olabilir.
MtDNA mutasyonlannın oftalmolojik belirtileri göz kapaklan, kornea ve ekstraoküler kaslardan oksipital kortekse kadar görme ekseninin hemen hemen tamamını kapsadığından dolayı belirgindir. Gözlerle ilgili belli başlı bulgular oftalrnopleji, optik nöropati ve pigmenter retinopatiyi içerir. Kardiyovasküler belirtiler dilate hipertrofık kardiyomiyopati, iletim hastalığı (conduction disease) ve kalp krizi, Wolff-Parkinson-White sendromu ve hipertansiyondur. Diabetes mellitus (şeker hastalığı) prevalansı mitokondriyal ensefalomiyopatili hasatalarda beklenenden daha yüksektir ve değişik mtDNA mutasyonlan ile birlikte ortaya çıkar. Diabetes mellitus çoğunlukla mtDNA daki 3243 nokta mutasyonuyla ilişkilidir, fakat sadece onunla değil sensorinöral duyma kaybı ile de olabilir.
YAYGIN HASTALIKLARDA MİTOKONDRİYAL DNA MUTASYONLARININ ROLÜ
Yaygın, sosyoekonomik önemli hastalıklarda mtDNA mutasyonlannın rolü aktif olarak araştırılmaktadır. Çoğu yaygın hastalığın genetik temeli komplekstir ve basit, tek genli Mendel kalıtımı göstermez. LHON ve aminoglikozid ile uyanlan sağırlık gibi mitokondri hastalıktan genetik ve epigenetik faktörler arasında kompleks fizyopatolojik etkileşim potansiyeli gösterir. Bu etkileşimler sonucunda, mtDNA mutasyonlan maternal kalıtım biçimi çok açık olmayan Diabetes mellitus gibi yaygın hastalıklann alt grubuna katılabilir.
Dokuya özgü somatik (kalıtsal olmayan) mtDNA mutasyonlarının birikimi Alzheimer ve Parkinson hastalığı gibi geç ortaya çıkan dejeneratif hastalıklarla muhtemelen ilişkilidir. Örneğin, insanlar yaşlandıkça bazal ganglia ve serebral korteks gibi mitoz geçirmeyen bazı dokular da dahil, dokularında mtDNA mutasyonlarının biriktiği gösterilmiştir. mtDNA daki yüksek mutasyon oranı ve zayıf tamir kapasitesi mitoz yapmayan dokularda veya daha yavaş geriye dönüşüm oranı olan dokularda, mtDNA mutasyonlarının birikimine katkı yapar. Tekrarlayan iskemi ve yeniden perfüzyon olması evrelerinde oluşan oksidatif hasar mtDNA “mutasyonlarının birikimini dikkate değer derecede artırır. Çevre faktörleri de mtDNA yı etkileyebilir. Retrovirüslere karşı kullanılan azidotimidin ilacı, kas mtDNA sini yok eder ve mitokondriyal miyopati oluşumuna neden olur. Topluca, mtDNA ve diğer mitokondriyal makromoleküllere önemli derecede oksidatif hasar ile ortaya çıkan bağlı mitokondri disfonksiyonu yaşlanmaya da katkı yapan etkendir. Mitokondriyal hastalıkarın tanısının moleküler genetik yöntemlerle kesin olarak yapılması, uygun genetik danışma ve en sonunda tedavi için gereklidir.