Anatomi ve Fizyoloji
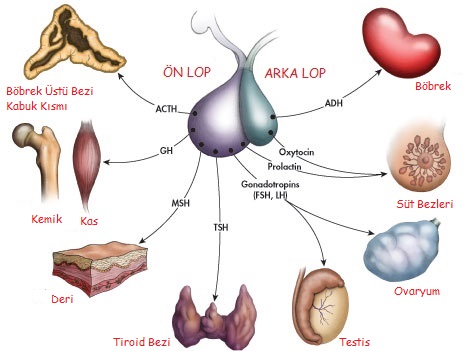
Hipofiz bezi 500-900 mg ağırlığında olup sphenoid kemikte bulunan sella tursika içinde kafa tabanında yer almaktadır. Kavernöz sinüsler her iki lateralde hipofiz bezinin sınırlarını oluştururlar. Bezin süperior kısmında optik kiyazma yer alır. Hipofiz bezinin üçte ikilik kısmını ön lob, üçte birlik kısmını ise arka lob oluşturur. Ön hipofiz bezini çoğunlukla hipotalamustan olmak üzere, hipotalamik-hipofizer portal dolaşım aracılığı ile, zengin bir vasküler ağ beslemektedir. Hipotalamik uyarıcı (stimülatör) ve baskılayıcı (inhibitör) hormonlar, hipofizer trofik hormonların sentez ve salgılanmasını kontrol etmek üzere, hipotalamik-hipofizer portal dolaşım aracılığı ile ön hipofiz bezindeki hücrelere direkt olarak ulaştırılır. Ön hipofiz hormonlarının her biri özelleşmiş ön hipofiz hücreleri tarafından salgılanırlar. Adrenokortikotrof hormon (ACTH), growth hormon (GH), prolaktin (PRL) ve tiroid stimüle edici hormon (TSH). Ek olarak gonadotrofinler, luteinize edici hormon (LH) ve folikül stimüle edici hormon (FSH), aynı hücre tarafından salgılanırlar. Arginin vazopressin (AVP) ise, antidiüretik hormon (ADH) olarak da adlandırılır, hipotalamusta sentez edilir ve hipofiz sapı (stalk) aracılığıyla depolanıp uyarı sonrası salgılandığı hipofiz arka lobuna ulaştırılır . Oksitosin ise aynı şekilde arka hipofiz lobunda depolanıp salgılanır.
Hipofiz Tümörleri
Hipofizde beş hücre tipinden, tek başına veya kombine olmak üzere, kendilerine özgü karakteristik hormonların salgılandığı benign hipofiz adenomlan gelişebilir. Sekretuar (hormon salgılayan) hipofiz tümörlerinden en sık görüleni prolaktinomalardır. Uzak metastaz yapan hipofiz karsinomları izole vakalar olarak rapor edilmiştir. Hipofiz tümörlerinin en erken klinik bulguları sıklıkla hormon aşırı salgılanmasına bağlı gelişen karakteristik bulgu ve belirtilerdir. Ayrıca tümör büyük ise kitledeki genişleme sonucu lokal etkiler de görülebilir. Etraf dokulara baskı büyük hipofiz adenomlarında (hormon salgılayan veya salgılamayan) belirti ve bulgulara neden olur. Baş ağrısı sık görülen bir belirtidir. Eğer tümör suprasellar alana ulaşmışsa optik kiazma bası altında kalır ve bu durumun klasik sonucu olarak bitemporal hemianopia gelişir.
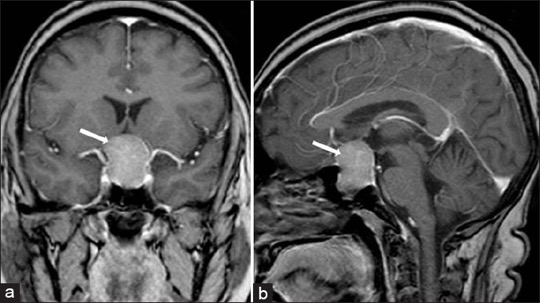
Lateralde kavernöz sinüs içine olan uzanım sonucu ise üçüncü, dördüncü ve altıncı kranial sinir disfonksiyonuna bağlı oftalmopleji, dipolopi veya pitozisa neden olabilirler. Büyüyen hipofiz tümörünü çevresindeki normal hipofiz dokusuna bası nedeniyle bir veya daha fazla hipofizer trofik hormonun salınımının azalmasına yani hipofiz yetmezliği (hipopituitarizm) bulgu ve belirtilerinin gelişmesine neden olabilirler. Destrüktif hipofizer lezyonlar belirli bir şekli izleyerek hormon eksikliği yapar. Öncelikle GH azalır, daha sonra LH ve FSH, sonra TSH takip eder, ve en son ACTH azalır. Hipofiz tümörünün varlığı magnetik rezonans görüntüleme (MRI) saptanır. Bir kontrast ajanı olan gadolinium görüntüleme sırasında normal hipofiz dokusundan küçük lezyonları ayırmada yardımcı olur. 10 mm’den küçük hipofiz lezyonları mikroadenom, 10 mm’den büyük lezyonlar ise makroadenom olarak tanımlanırlar. Hipofiz adenomları hipofiz sapının anlamlı bir şekilde karşı tarafa sapmasına neden olabilirler. Boyutu 15 mm’yi aşan adenomlar sıklıkla suprasellar uzanım gösterirler ve optik kiyazmada kompresyona neden olurlar.
MRI aynı zamanda büyük adenomların kavernöz sinüs içine olan lateral uzanımlarını da gösterebilir. Genişlemiş sellanın en sık nedeni olan empty sella (boş sella) sendromuna, primer konjenital defekt veya cerrahi/radyasyon sonrası gelişen inkomplet diyafragma selladan araknoid membranın herniasyonu neden olabilir. Endokrinolojik değerlendirme her zaman görüntüleme çalışmalarından önce yapılmalıdır. Çünkü toplumun yaklaşık %l0-20’sinde MRl ile tesadüfen (insidental olarak) saptanan işlevsel olmayan hipofiz mikroadenomları saptanabilir.
Ön Hipofiz Hormonlarının Bozuklukları
GROWTH HORMON
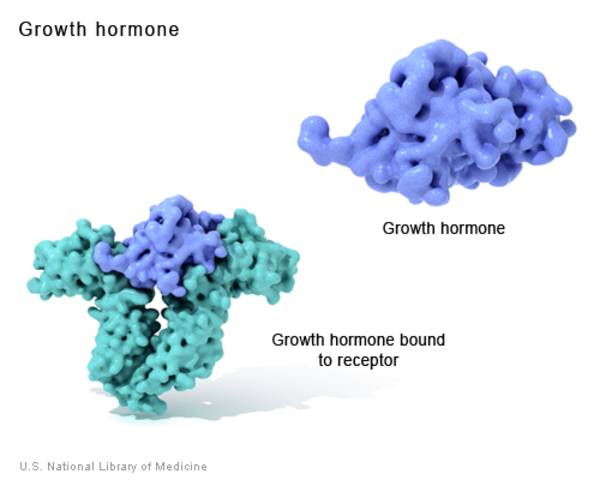
GH 191 aminoasitten oluşan ve 22,000 Dalton ağırlığında bir peptit hormondur. Sekresyonu 40 ve 44 amino asitten oluşan hipotalamik growth hormon-salgılatıcı hormon (GHRH ) ile uyarılır ve hipotalamik tetradekapeptid somatostatin tarafından baskılanır. Bu hipotalamik faktörler hipofizdeki somatotrof hücrelere bağlanarak GH salınımını düzenlerler. GH karaciğerde reseptörüne bağlanır ve insülin benzeri growth faktör-1 (IGF-I) salınımını uyarır. IGF-I dolaşımda, en önemlisi IGF-BP3 olmak üzere, bağlayıcı proteinlere (BPs) bağlanarak taşınırlar. IGF-I GH’ın büyüme uyarıcı etkilerinin çoğuna aracılık eder. GH aynı zamanda karbonhidrat metabolizmasında etkilidir.
GH eksikliğini değerlendirebilmek için somatotof hücreleri uyaran provokatif testler gereklidir çünkü bazal GH düzeyleri normal bireylerde de sıklıkla çok düşük düzeydedir. İnsüline bağlı hipoglisemi (İTT: insülin tolerans testi) GH rezervini değerlendirmek için altın standart testtir. İnsülin (0.05-0.15 U/kg), hastanın kan şeker düzeyi başlangıcın %50’si veya 40 mgldl’nin altına düşürmek üzere, intravenöz olarak uygulanır. Hipoglisemi GH salınması için potent bir uyarıdır ve normal cevap ise pik GH düzeyinin 60. dakikada 5ng/mL’nin üzerine çıkmasıdır. GHRB ve argininin kombine olarak infüzyonu da erişkinde GH salınımını uyarmada İTT kadar duyarlı ve özgüldür. Oral propranolol ve L-dopa. dopamin ve norepinefrinin öncüsü, hipofizer somatotrof hücrelerden GH salınmasını uyarırlar ve çocukluk dönemi GH eksikliğinin değerlendirilmesinde kullanılırlar, fakat erişkinde GH eksikliği tanısında duyarlı değillerdir. Bir hastada GB eksikliği tanısı koyabilmek için birden fazla test yapılmalıdır çünkü her bir teste normal bireylerin
yeterli cevap verme oranı sadece %90 kadardır. IGF-l GH tarafından kontrol edildiği için IGF-l düzeyleri GH eksikliği için tarama testi olarak kullanılabilir. GH eksikliği düşük IGF-I düzeyi ile ilişkilidir. Yani düşük IGF-I düzeyleri GH salınmasının, provakatif test uygulanarak kontrol edilmesi gereğini doğurur.
GH hipersekresyonunun değerlendirilmesi daha kolaydır. GH pulsatil olarak salgılanır ve böylelikle herhangi bir zamanda yapılan tek GH ölçümüne göre dinamik GH testi çok daha değerlidir. Ayrıca, siroz, açlık, anksiyete, tip 1 diyabetes mellitus, ve akut hastalık gibi durumlar GH hipersekresyonuna neden olabilirler. Ancak serum IGF-I düzeyleri gün boyunca dalgalanma göstermediği için GH hipersekresyonunu göstermede faydalı bir belirteçtir. GH hipersekresyonu olan hastaların hemen hepsinde IGF-I düzeyleri yükselir. GH hipersekresyonunu göstermek için basit ve spesifik bir dinamik test olan oral glukoz uygulanması, sağlıklı bireylerde 100 gr oral glukozun 120 dakika sonra GH düzeylerini 1 ng/mL’nin altına baskılaması esasına dayanır. Akromegalik hastalarda oral glukoz yüklemesinden sonra GH düzeyleri yükselebilir, değişmeden kalabilir veya azalabilir (ancak 1 nglmL’nin altına düşmez). Eğer hipofızer bir odak bulunamazsa, göğüs ve abdominal görüntüleme çalışmaları ile ektopik GH veya GHRH salgılayan ekstrahipofızer bir kaynak aranmalıdır.
Bebeklik ve çocukluk döneminde GH eksikliği gelişme geriliği ve açlık hipoglisemisi ile kendini gösterir. Erişkin GH eksikliğinde ise görülen temel tablo, artmış abdominal obezite, azalmış kas kuvveti ve egzersiz kapasitesi, azalmış yağsız vücut kütlesi ve artmış yağ kütlesi, azalmış kemik mineral yoğunluğu, glukoz intoleransı ve insülin direnci, anormal lipid profili, ve bozulmuş psikososyal iyilik halidir. Erişkinde büyüme hormon eksikliğine sıklıkla hipofiz yetmezliğinin diğer semptomları eşlik eder. Tedavide erişkinde GH günlük (0.1-0.3 mg/gün) subkutan enjeksiyonlar halinde uygulanır. GH tedavisi sonrasında erişkinlerde yağ kütlesi azalır, yağsız vücut kütlesi ve kemik mineral yoğunluğu artar, ve GH replasman tedavisi
kardiyovasküler risk faktörlerinde iyileşme ile ilişkilidir.
Akromegali ve Gigantizm (Devlik)

Çocukluk döneminde GH hipersekresyonu gigantizme yol açar; uzun kemiklerde epitizlerin kapandığı erişkinlerde ise GH fazlalığı akral bölgelerdeki lokal büyüme ile karakterize akromegaliye neden olur. GH hipersekresyonu hemen her zaman GH salgılayan hipofiz adenomu nedeniyle olur. Akromegalik hastaların yaklaşık %70’inde tümörler 1 cm’nin üzerindedir. Ektopik GHRH salınımı pankreatik adacık hücreli tümörlerde, ve bronşial ve intestinal karsinoidlerde görülebilir. Ektopik GH salınımı çok nadir olarak pankreas, meme ve akciğer tümörlerinde görülebilir. Hem ektopik GH hem de ektopik GHRH klinik olarak kendini akromegali olarak gösterir ancak ileri derecede nadir görüıürler.
Akromegalinin klinik bulguları sessizdir ve şekil değişikliklerinin fark edilip tanı konması yıllar alabilir. Tedavi edilmemiş akromegali, hastalığın ilerleyen yıllarında artmış morbidite ve mortaliteye neden olur. En klasik klinik bulgu ellerin ve ayakların genişlemesi ve yüz hatlarının kabalaşması ile karakterize akral (uç) büyümedir. Ayrıca frontal sinüslerde genişleme, supraorbital kenarların belirginleşmesi ve alt çenenin aşağı ve öne büyümesi sonucu prognatizm ve dişlerin arasında açılma görülür. Eller ve ayaklarda yumuşak doku genişlemesine bağlı yüzük, eldiven ve ayakkabı numaraları artar. Kemik ve yumuşak dokudaki değişikliklere endokJin, metabolik ve sistemik olaylar da eşlik eder.
Tedavide ise transsfenoidal mikrocerrahi ilk tedavi seçeneğidir, çünkü GH düzeylerinin hızlı düşmesi ve düşük orandaki cerrahi morbidite görülür. Kür oranları preoperatif tümör boyutları ile orantılı olmakla birlikte mikro adenomlarda %90’lara ulaşmaktadır. GH hipersekresyonunu azaltmak için radyoterapi etkin bir metod olmakla birlikte radyoterapi sonrası GH düzeylerinin düşmesi için geçen süre 20 yılı bulabilir ve hipofiz yetmezliği insidansı da yüksektir. Tıbbi yaklaşım ise GH salınımının hipofiz tümöründen salınımının azaltılması (somatostatin analogları, dopamin agonistleri) veya GH’nun karaciğer reseptöründeki periferal etkisinin bloke edilmesi (GH-reseptör antagonistleri) şeklindedir. Oktreotid asetat, uzun etkili somatostatin analoğu, GH ve TGF-T düzeylerini normale indirmede hastaların %40-%65’inde etkilidir ve bazı vakalarda tümör kitlesi belirgin olarak küçülür.
Oktreotidin aylık yapılan ve uzun etkili, yavaş salınımlı depo preperatı, kısa etkili subkutan oktreotid kadar etkilidir. Oktreotidin yan etkileri, ishal, abdominal kramplar, şişkinlik, ve safra taşı oluşumudur. Bir dopamin agonisti olan bromokriptin sadece az bir kısım akromegalik hastada GH baskılama yönünden etkilidir. GH reseptör antagonisti, pegvisomant, hepatik yüzeydeki GH reseptörüne bağlanır GH’nun normal fizyolojik etkilerini bloke eder. GH’nun karaciğerdeki fizyolojik etkisinin mekanizması; GH’nun hepatik reseptöre bağlanması, sonrasında reseptör dimerizasyonunun IGF-I oluşması için sinyal yolaklarını aktive etmesi, aşamalarını içerir. Pegvisomant GH-reseptör dimerizasyonunu ve dolayısıyla lGF-1 oluşumunu bloke eder ve TGF-Tdüzeylerini akromegalik hastaların %97’sinde azaltır. Karaciğer fonksiyon testleri ve hipofiz adenom boyutları uzun dönemde takip edilmelidir.
PROLAKTİN
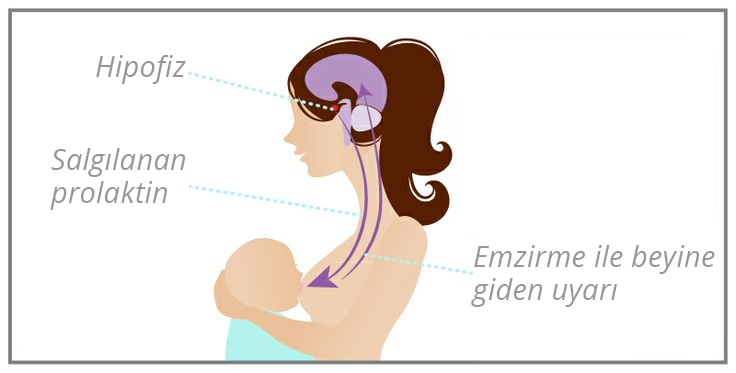
PRL, 198 amino asitten oluşan 22.000 Da polipeptid, hipofizer latotrof hücreler tarafından sentez edilir ve salınır. Hipotalamik dopamin PRL salınımı üzerine baskın bir inhibitör kontrol sağlar ve sadece düşük düzeyde bir bazal salınıma neden olur. Tirotropin salgılatıcı hormon (TRH) ve vazoaktif intestinal polipeptid bilinen PRLsalgılatıcı faktörlerdir. PRL salınımı dönemsel bir şekilde olur. Estrojenler bazal ve uyarılmış PRL salınımı artırırlar; glukokortikoidler ve TSH ise TRH bağımlı PRL salınımı baskılarlar. Gebelikte PRL düzeyleri artar ve doğum sonrasında PRL süt üretimini uyarıro Ancak laktasyonun devamlılığı için PRL düzeyleri gerekli değildir, ve bazal PRL salınması doğum sonunda düşer ve laktasyonu bebeğin emme refleksi devam ettirir.
Prolaktinomalar
Mikroprolaktinomalar kadınlarda daha sık olmakla birlikte, makroprolaktinomalar sıklıkla erkeklerde görülür. PRL pulsatil gonadotropin baskılar ve siklus ortasındaki LH artışını da baskılayarak menstrual düzensizliklere neden olur. Kadında hiperprolaktinemi hipogonadotropik hipogonadizme ve bunun sonucu olarak da östrojen eksikliğine neden olur. Hiperprolaktinemisi olan erkekler testosteron düzeyleri genellikle baskılıdır.
Klinik bulgulara bakarsak prolaktinomalar menstrual düzensizlik ve infertilite olan kadınlarda, libido azlığı ve impotans olan erkeklere göre daha erken fark edilirler. Hiperprolaktinemisi olan kadınların yaklaşık %90’ında amenore, galaktore ve infertilite görülür. Eğer prolaktinoma menarş başlamadan önce adolesan yaşta ortaya çıkarsa primer amenore görülebilir. Prolaktinomalar sekonder amenore nedenlerinin %15-%20 kadarını oluşturur. Anovulasyon infertilite ile ilişkilidir. Galaktore klinik olarak aşikar olmayabilir ve meme muayenesi sırasında fark edilebilir. Östrojen eksikliği osteopeni, vajinal kuruluk, sıcak basmaları, ve irritabiliteye neden olabilir. PRL adrenal androjen üretimini uyarabilir ve bu durum sonucunda kilo artışı ve hirşutizm gelişebilir. Hiperprolaktinemi anksiyete ve depresyon ile de ilişkili olabilir. Erkekler hipogonadizm sonucu libido kaybı ve impotans ile başvururlar. Bu semptomlar genelde prolaktinoma dışındaki nedenlere bağlanır, ve görme alan kaybı, baş ağrısı ve hipofiz yetmezliği gelişene kadar sıklıkla tanıda gecikmeye neden olur.
Tanı PRL salınmasını, çeşitli fizyolojik durumlar (gebelik, stres, meme başı uyarısı gibi), bazı grup ilaçlar (fenotiyazinler, metildopa, simetidin, metoklopramid gibi) ve patolojik durumlar (hipotiroidizm, kronik böbrek yetmezliği, göğüs duvarı lezyonları gibi) artırabilir. Bu bahsedilen durumlar genellikle hafif derecede PRL yükselmesine neden olurlar. Ancak 200 ng/ml’nin üzerindeki PRL düzeyleri genellikle prolaktinomayı düşündürür. MRI ile prolaktinoma tanısı onaylanmalıdır.
Tedavide ise dopamjn agonistleri ile tıbbi tedavi (bromokriptin, kabergolin) gonadal işlevleri ve fertiliteyi hastaların çoğunda düzeltir. Dopamin agonistleri makroprolaktinoması olan önemli sayıdaki hastada tümör küçülmesine neden olurlar. Transsfenoidal cerrahi medikal tedaviyi tolere edemeyen ve dirençli hastalarda endikedir.
TİROİD-STİMÜLAN (UYARICI) HORMON (TSH)
TSH, 28.000 Dalton ağırlığında bir glukoprotein hormondur ve hipofizde tirotrof hücreler tarafından sentezlenir ve salınır. TSH sekresyonu hipotalamik bir tripeptid olan TRH tarafından uyarılır. Hipotalamik somatostatin, periferik tiroid hormonlarının TSH salınması üzerindeki negatif feedback etkisini artırır. TSH tiroid bez i üzerindeki reseptörüne bağlanarak adenilat siklazı aktive eder, iyod alımını ve tiroksin (T4) ve triiodotironin (T3) sentez ve salınmasını uyarır. T4 ve T3, hipofizer TSH ve hipotalamik TRH salınması üzerinde negatif feedback etki gösterir.
TSH salınmasının değerlendirilmesi ultrasensitif metodlarla (immünoradyometrik) ölçülmektedir ve düşük, normal ve yüksek TSH düzeyleri doğru bir şekilde ayrılabilmektedir. Ultrasensitif TSH ölçümleri ileri dinamik testlerin yerini büyük ölçüde almıştır. Hipotiroidizmi olan hastada, baskılı TSH düzeyleri santral (sekonder) hipotiroidizmi, yüksek TSH düzeyleri de primer hipotiroidizmi düşündürür.
TSH eksikliği, tiroid bezi involüsyonuna, hipofonksiyonuna ve hipotiroidizm kliniğine neden olur. Hipotiroidizmin klinik belirtileri, letaıji, kabızlık, soğuk intoleransı, bradikardi, ağırlık artışı, azalmış iştah, kuru deri, ve periferal reflekslerde relaksasyon zamanında gecikme olarak sıralanabilir. Dolaşımdaki TSH düzeyinin tiroid hormon düzeyleri ile birlikte düşük olması, hipotalamik-hipofizer işlevsizlik sonucu gelişen ikincil hipotiroidizmi birincil hipotiroidizmden ayırır.
Tedavi de ise santral hipotiroidizmi olan hastalar 75-150 mcg/gün dozlarında tiroksin ile tedavi edilirler. Tiroid replasmanının yeterliliği sT4 ölçümü ve bu değerin ortanormal aralıkta olması ile değerlendirilir. Santral hipotroidizmde TSH düzeyleri düşük-normal aralıkladır; böylelikle TSH düzeyleri santral hipotiroidizmde tedaviyi izlemek için yeterli değildir.
Tirotropin (TSH) salan tümörler oldukça nadirdir. Hipertiroidizm, guatr ve yüksek serum tiroid hormon düzeylerine rağmen uygunsuz olarak yüksek (veya normal) TSH düzeyleri ile kendini gösterir. TSH salan eden tümörler genelde tümörler genellikle plurihormonaldir ve TSH yanında GH, PRL ve glukoprotein hormon alfa alt birimini de salarlar . Bu tümörler cerrahiye dirençlidir ve birden fazla cerrahi girişim veya radyoterapi gerekebilir. Oktreotid asetatın, somatostatin analoğu, bazı hastalarda TSH salınmasını azalttığı ve tümör kitlesini de küçülttüğü gösterilmiştir. Tirotoksikozu kontrol etmek için iyod-131 ablasyonu veya tiroid cerrahisi gerekli olabilir.
ADRENOKORTİKOTROFHORMON (ACTH)

ACTH, 39 aminositten oluşan peptid, 241 aminoasitlik büyük bir ön molekülün parçası olarak sentez edilir. Ön molekül olan proopiomelanokortin enzimatik olarak anterior hipofiz lobunda beta-lipotropin , ACTH, bağlayıcı peptid, ve amjno terminal peptid şeklinde parçalanır. ACTH daha sonra beta-melanosit stimüle eden hormon ve kortkotropin benzeri peptid B-LPH ise LPH ve B-endorfin olarak ayrılırlar. Hipotalamik kortikotropin-salgılatıcı hormon (CRH) ve daha az olarak da ADH, hipofizer kortikotrof hücrelerden ACTH salınmasını uyarıro ACTH adrenal bezde kortizol sentez ve salınmasını uyarır. Kortizol ACTH ve CRH salınması üzerine negatif-feed- L back etki gösterir. ACTH pulslar halinde salgılanır ve uyanmadan önceki son saatlerde maksimuma ulaşan ve öğleden sonra sabit olarak en alt düzeye düşen sirkadian kontrol altındadır. Psikolojik ve fiziksel stres ACTH ve kortizol salınmasını artırır, ancak kortizol ACTH sekresyonunu, ve CRH ve ADH sentez ve salınmasını inhibe eder. ACTH aynı zamanda protein sentezini artırarak adrenal boyutları korur.
ACTH’nın fazla miktarda salınması lliperkortizolemi ile sonuçlanır, ve bu durum ACTH salan eden hipofız adenomu (Cushing hastalığı) veya ektopik ACTH salınması nedeniyle olur. ACTH eksikliği azalmış kortizol salınması ile birlikte adrenokortikal yetmezliğe neden olur. Aldosteron salınması, büyük oranda renin anjiyotensin aksı ile düzenlendiği için genellikle normal kalır. Bazal ACTH düzeylerİ. ACTH düzeylerinin rastgele ölçümü kısa plazma yarılanma ömrü ve pulsatil salınması nedeniyle güvenilir değildir. Plazma ACTH düzeylerinin yorumlanması için eşzamanlı kortizol düzeyleri ölçülmelidir. ACTH kortizol salınmasını düzenlediği için plazma kortizol düzeyleri hipotalamo-hipofızer-adrenal aksı daha iyi yansıtır. Sabah 8 kortizol düzeyinin 3 mcg/dl’nin altında olması adrenal yetmezliği düşündürür. ACTH düzeyleri primer adrenal yetmezliği sekonderden ayırmak için kullanılabilir. Primer adrenal hastalıklara bağlı adrenal yetmezliklerde plazma ACTH düzeyleri normal veya yüksek ölçülür, hipotalamo-hipofizer hipofonksiyonuna bağlı adrenal yetmezliklerde ise düşük ölçülür veya ölçülemez düzeydedir .
ACTH rezervinin yeterliliğini değerlendirmek için provokatif testler yapılmalıdır. Daha önce hipofizer GH salınması için uyaran olarak tanımlanan insüline bağlı llipoglisemi testi aynı zamanda hipotalamo hipofizer adrenal aksı da uyanrır. Pik kortizol düzeyinin en az 18 mcg/dl’nin üzerinde olması normal ACTH rezervini gösterir. Stres sonrası ACTH cevabında insülin hipoglisemi testi en güvenilir testtir. Ancak bu test yaşlı hastalarda ve serebrovasküler hastalığı, epilepsi veya kardiyovasküler hastalığı olanlarda kontrendikedir. Metirapon (30 mg/kg gece yarısı oral yoldan verilir) adrenal bezde 11-deoksikortizolü kortizole çeviren 11-beta hidroksilaz enzimini inhibe eder.
Düşük kortizol CRH ve ACTH’yl uyarır. Metirapon uygulanmasından sonraki sabah 11-deoksikortizol düzeyi 7 mg/dl’den fazla ise, eş zamanlı kortizol ölçümü 5mcg/dl’den küçük olmalıdır, yeterli cevap olarak kabul edilir. Eğer baskılanmış adrenal fonksiyon düşünülüyorsa metirapon ve ITT potansiyel olarak zararlı olabilir ve hasta doktoru tarafından yakından monitorize edilmelidir.
Uzun süren ACTH eksikliği adrenal atrofiye neden olur, böylelikle adrenal kortizal rezerv ölçülerek ACTH’nın durumu dolaylı yoldan değerlendirilebilir. ACTH uyarı testi ile ITT arasında iyi bir korelasyon vardır. Yaklaşık olarak 250 mcg cosyntropin intravenöz veya intramusküler olarak uygulanır ve normal bireylerde 60 dakika içinde pik kortizol cevabı 20 mcg/dL’den yüksek olur.
Cushing hastalığında hipofizer kortikotrof adenom veya ektopik ACTH salan tümörler ACTH hipersekresyonuna ve hiperkortizolemiye neden olular. Öncelikle 24 saatlik serbest kortizol düzeyini ölçerek, gece yarısı plazma kortizol düzeyi, veya gece 11.00 de alınan img deksametazon sonrası (lmg gecelik deksametazon süpresyon (baskılama testi) sabah 8.00 de serum kortizolünün baskılanmaması ile, hiperkortizolemi tanısı onaylanır. Cushing sendromu ve hiperkortizolemi onaylandıktan sonra, ACTH bağımlı Cushing sendromu (hipofizer veya ektopik ACTH salınımı) veya ACTH bağımsız Cushing sendromunu (adrenal adenom ve hiperplazi) ayırmak için plazma ACTH düzeyi ölçülür.
Adrenal yetmezlikle sonuçlanan ACTH eksikliği, letarji, yorgunluk, bulantı kusma, diyare, ortostatik hipotansiyon, koma ve tedavi edilmezse ölüme neden olabilir. Adrenal yetmezliği olan hastalar 15-25 mg/gün bölünmüş dozlarda hidrokortizon veya eşdeğeri bir kortikosteroid ile tedavi edilir. Toplam dozun üçte ikisi sabah üçte biri ise öğleden sonra veya akşam verilir. Stres durumlarında bu doz 5-10 kat artırılır. Eğer hasta operasyona gidecek ise stres dozlar uygulanır ve operasyon sonrasında hızla doz azaltılır. Her hastada hafif bir hastalık mevcutsa hidrokortizon dozu iki kat artırılır. Santral adrenal yetmezliğinde renin aldosteron sistemi sağlam olduğu için mineralokortikoid replasmanı gerekmemektedir.
ACTH salan hipofiz tümörleri hipokortizolemiye ve bununla ilişkili olarak da obezite, ay dede yüz, servikodorsal displazi, stria, ciltte incelme, hirşutizm, hipertansiyon, adet düzensizlikleri, glukoz intoleransı, davranış değişiklikleri, proksimal mjyopati, ve osteopeni görülür. Tedavide cushing hastalığında ACTH hipersekresyonunun kontrolü için kullanılan tedavi modaliteleri şu şekildedir; transsfenoidal cerrahi, radyasyon tedavisi ve medikal tedavi. Steroid sentezi bloke eden ilaçlar ketokenazol, metirapon ve aminoglutetimid, RU-486, ve trilostan gibi ilaçları içerir. Ektopik ACTH sendromunun tedavisi mümkünse ektopik tümörün çıkarılması ile olur.
GONADOTROPİNLER (LH VE FSH)

Gonadotropinlerin (LH ve FSH) salınması, pulsatil olarak salınan ve 10 aminoasitten oluşan bir peptid olan hipotalamik gonadotropin salgılatıcı hormon (GnRH) tarafından düzenlenir. Gonadal steroidler (estrojen ve testosteron) ve peptidler (inhibin ve aktivin) negatif geri besleme inhibisyonunu düzenler. Bazal LH ve FSH pulsatil olarak salınırlar ve bu salınım GnRH’nın pulsatil salınması ile uyumludur. GnRH salınımı pubertenin başlangıcını ve ovulasyon için gerekli olan siklus ortasındaki gonadotropin artışını belirler. Gonadal steroidler gonadotropin salınması üzerine hem pozitif hem de negatif geri besleme etkisine sahiptir. Ayrıca gonadal polipeptid olan, ve overlerde granüloza hücreleri ve testislerde Sertoli hücreleri tarafından üretilen inhibin, FSH salınmasını negatif olarak inhibe ederken, aktivinler ise FSH salınmasını uyarırır.
LH ve FSH over ve testislerdek reseptörlerine bağlanır ve seks steroid salınması (özellikle LH), ve gametogenezi (özellikle FSH) uyarırlar. LH gonadal stroid salınması testisin Leydig hücresi ve over follikülleri aracığı ile yapar. Kadınlara ovulatuar LH piki follikülün yırtılmasına ve lüteinizasyona neden olur. FSH erkekte Sertoli hücrede spermatogenezi ve kadında ise follikül gelişmesini sağlar.
Hastalarda LH ve FSH düzeyleri yaşla değişir ve kadınlarda ise menstruel siklus ile değişir. Püberte öncesi gonadotropin düzeyleri düşüktür, ve postmenopozal kadınlarda artmış düzeyler mevcuttur. Erkeklerde FSH ve LH düzeyleri pulsati! olmakla birlikte kadınlardakinden daha az dalgalanma gösterir. Menstruel siklusun folliküler fazında LH düzeyleri istikrarlı bir şekilde artar ve siklus ortasındaki piki ovulasyonu uyarır erken folliküler faz süresince FSH düzeyi artar, geç folliküler fazda ise düşer, ve siklus ortasında LH piki ile uyumlu olarak pik yapar. Ovulasyon sonrası LH ve FSH düzeyleri düşer. Erkeklerde normal pulsatil salınmayı yansıtabiIrnek için LH ve FSH düzeyleri 20 dakika aralarla alınan üç örneğin havuzlanması şeklinde ölçülür.
Gonadotropin ve seks steroid hesaplanması kadınlarda çok daha komplekstir. Ancak regüler menstrüel siklusu olan ve luteal fazda serum progesteron konsantrasyonu normalolan kadınlarda gonadotropin işlevsizlik ihtimali çok düşüktür. Amenore olan bir kadında serum LH, FSH, estradiol, PRL,ve hUlnan korionik gonadotropin (hCG) ölçülerek şu tanılar birbirinden ayrılır:
(1) primer ovaryan yetmezlik, artmış FSH ve LH düzeyleri ve normal PRL düzeyleri olur;
(2) hiperprolaktinemi, artmış PRL ve normal-düşük folliküler faz LH,FSH ve estradiol düzeyleri;
(3) gebelik, pozitif hCG, normal-yüksek PRL, normal LH, ve yüksek estradiol olur.
Gonadotropin eksikliğinin tanısı en iyi serum gonadotropinleri ve gonadal steroidlerinin eş zamanlı olarak birlikte ölçülmesiyle konur. Düşük-normal FSH ve LH düzeyleri ile birlikte düşük testosteron düzeyleri (erkeklerde) veya düşük estradiol düzeyleri (kadınlarda) gonadotropin eksikliği tanısını onaylar. Artmış gonadotropin düzeyleri ile birlikte düşük düzeylerde gonadal steroidlerin olması primer gonadal yetmezliği
düşündürür.
Gonadotropin eksikliği çocukluk dönemindeki santral hipogonadizim normal pubertal gelişmenin olmamasıyla sonuçlanır. Kızlarda gecikmiş meme gelişimi, pubik ve aksiler kıllarda azalma, ve primer amenore görülür. Erkeklerde ise, penil ve testiküler gelişme olmaz, ve vücut kılları belirgin olarak azdır. Uzun kemiklerde epifizlerin kapanması için seks steroidleri gereklidir; böylelikle izole gonadotropin eksikliğinde, epifizyal füzyon olmadığı için büyüme devam eder (GH salınması normal ise) ve bu durum adölesanlarda enükoid oranların olduğu bir boy uzamasına neden olur. Erişkin kadında, hipogonadizm kendini meme atrofisi, pubik ve aksiler kılların kaybı, ve sekonder amenore ile kendini gösterir.
Hipogonadal erişkin erkekteki oluşan değişiklikler ise; tetstiküler atrofi, libidoda azalma, impotans, kas ve kemik kütlesinde azalma, vücut kıllarının kaybı, ve artmış LDL kolesterol düzeyleridir.
Tedavisinde santral hipogonadizmli erkeklerde androjen replasmanı (yerine koyma tedavisi) önerilmektedir. Testosteron intramusküler enjeksiyon olarak (testosterone enanthanate 200-300 mg her 2 veya 3 haftada bir kez) veya transdermal olarak (2.5-5 mg/gün) veya jel olarak (2.5-10 mg/gün) uygulanabilir. Yeterli replasman tedavisi normal aralığın orta düzeylerinde bir serum testesteron düzeyi ile sonuçlanır. Santral hipogonadizmi olan premenapozal kadınlar ise estrojen tedavisi almalıdır, eğer uterusları mevcutsa, uterusu korumak için beraberinde sürekli veya siklik olarak progesteron tedavisi gereklidir. Postmenopozal hipofiz yetmezlikli kadınlarda estrajen progesteron replasmanı ile ilgili öneriler, hipofiz yetmezliği olmayan postmenopozal kadınlarla aynıdır.
Gonadotropin salgılayan hipofiz tümörleri nadirdir ve özellikle erkeklerde rapor edilmiştir. Tümörlerin önemli çoğunluğu tanı anında büyüktür ve sadece FSH hipersekresyonu olur. Hastalar genellikle görme bozukluğu gibi lokal basıya bağlı belirti ve bulgulara sahiptir. Hastalarda ayrıca düşük veya normal testosteron düzeylerinin veya düşük veya normal sperm sayılarının olduğu hipogonadizm görülebilir. Bu durumun nedeni kronik olarak yüksek düzeylerdeki (LH ve FSH) gonadotropinlerin testisler üzerindeki inhibitör etkileridir. Nadiren bazı vakalarda aşırı LH sahnımı testesteron düzeylerini artırabilir. Gonadotropin salgılayan eden adenomların cerrahi olarak çıkarılmaları primer tedavi seçeneğidir; ancak hastalar izleyen radyoterapiye de ihtiyaç duyarlar.
HİPOTALAMAİK İŞLEVSİZLİK
Çocuklar ve genç erişkinlerde kraniofarengioma hipotalamus işlevsizliğinin en sık nedenidir. Primer santral sinir sistemi tümörleri, pinealomlar, dermoid ve epidermoid tümörler de erişkinde hipotalamus işlevsizliğinin nedenleridir. Hipotalamus işlevsizliği yapan tümörlerin klinik belirtileri; görme kaybı, artmış intrakranial basınç belirtileri (başağrısı ve kusma), hipofiz yetmezliği (büyüme
geriliğini de içerir) ve diyabetes insipidus, olarak sıralanabilir. Hipotalamus değişiklikleri susama bozuklukları (poliüri, polidipsi, dehidratasyon), iştah bozuklukları (hiperfaji ve obezite), ısı düzenlenmesi ve şuur bozukluklarını (somnolans, duygusal değişiklikler) içerir. Diyabetes insipidus hipotalamus lezyonlarının sık bir bulgusu olmakla birlikte primer hipofiz lezyonları nadiren görülür. MR tanıyı ile tanı doğrulanır. Hipotalamus lezyonlarında hipofiz yetmezliği sık görüldüğü için ön hipofiz işlevleri araştırılmalıdır. Kraniofarengioma cerrahi rezeksiyon ile tedavi edilir ve izleyen dönemde radyoterapi gerekebilir. Diğer hipotalamus tümörlerinin cerrahi rezeksiyon öncesi histolojik tanısı için bazen biyopsi gerekebilir, çünkü disgerminom gibi bazı tümörler radyosensitif olabilir.
Hipofiz Yetmezliği (Hipopituitarizm)
Bir veya birden çok hipofiz hormonlarının azalması ile hipofiz yetmezliği gelişir. Bu sendrom ya ön hipofiz bezinin tahribine bağlı gelişir ya da normalde hipofizin işlevini düzenleyen hipotalamisun uyarıcı baskılayıcı etkenlerin eksikliğine ikincil olarak gelişir. Konjenital (doğumsal) veya sonradan gelişen lezyonlar da hipofiz yetmezliğine neden olabilirler. Hipofiz yetmezliği genellikle yavaş bir şekilde gelişir ve hipofiz lezyonları bir veya birden fazla hormon kaybına neden olurlar.
Tedavisinde panhipopituitarizmi (tam hipofiz yetmezliği) olan hastalar T4, glukokortikoid ve uygun seks steroidleri ile yeterli bir şekilde replasman (yerine koyma) tedavisi almalıdırlar. GH eksikliğine bağlı boy kısalığı olan çocuklar GH replasman tedavisi almalıdır. GH eksikliği olan erişkinlerde GH tedavisinin etkinliği ve güvenliği halen araştırılmaktadır. Erkeklerde testesteron tedavisi ile libido ve impotansı düzelir, vücut kıllarının büyümesi ve kas kuvveti de normale gelir. Kadınlarda estrojen tedavisi sekonder seks karakterinin devamını sağlar ve sıcak basmasını önler. İnsan menapozal gonadotropinlerin ve hCG’nin intramusküler olarak veya GnRH’nın infüzyon pompası ile verilmesi ovulasyonu sağlayabilir. TSH ve ACTH’nın birlikte eksik olduğu durumlarda, glukokortikoidler T4’den önce replase edilmelidir, çünkü T4 adrenal yetmezliği alevlendirebilir ve akut adrenal yetmezliğe neden olabilir.
Arka Hipofiz Bezi
Arka hipofiz bezi ADH ve oksitosin salar. Bu hormonlar, hipotalamustan arka hipofize uzanan hipotalamik nükleuslarda bulunan nöronal hücre gövdelerinde üretilirler. ADH renal tübüllerdeki reseptörlerine bağlanır ve toplayıcı kanal epitelinin luminal membranında su geçirgenliğini artırarak su geri emilimini uyarır ve idrarın konsantre edilmesini sağlar. En fazla düzeyde ADH’nın etkisi sonucu yüksek ozmolaritede (1200 mOs m/kg olacak kadar yüksek) az hacimde konsantre idrar oluşur. ADH eksikliği ise çok dilüe (100 mOsm/kg olacak kadar düşük) yüksek miktarda idrara neden olur. Böbrek tübüllerine olan etkilere ek olarak, ADH periferik arteriolar reseptörlere bağlanır ve damarların kasılmasına neden olarak kan basıncını yükseltir. Ancak bu hipertansif etkiye zıt bir etki olarak, ADH aynı zamanda bradikardi ve sempatik sinir sistemi aktivitesinde inhibisyona neden olur. ADH’nın eksikliği veya böbreklerin ADH’ya duyarsızlığı polüri, polidipsi ve noktüri ile giden diyabetes insipidusa neden olurlar. ADH’nın fazla miktarda ve uygunsuz olarak salgılanması uygunsuz antidiüretik hormon üretimi ile giden sendroma (SIADH) neden olur ve hiponatremik bir durum oluşur. Oksitosin uterusta düz kas kontraksiyonuna neden olur.
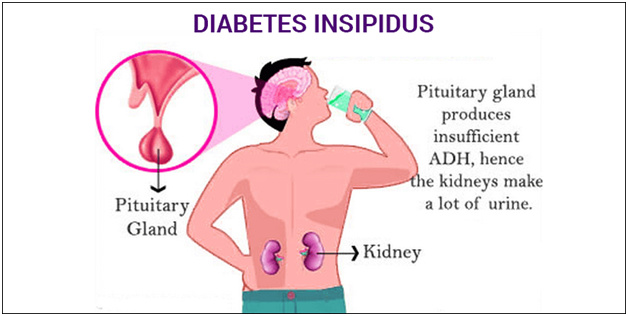
Arka hipofiz işlevsizliğinde vazopressin eksikliği görülür ve poliüri, polidipsi ve noktüri ile birlikte giden diyabetes insipidusa yol açar. Diyabetes insipidus, hipofizin arka lobunun yeterince ADH salamadığı santral (nörojenik) kökenli olabilir; veya böbreğin dolaşımda yeterli miktardaki ADH’ya cevapsızlığına bağlı olarak nefrojenik kökenli olabilir. Nedenden bağımsız olarak hastalar poliüriktir ve aşırı miktarda dilüe idrar çıkarırlar. Bu durum hücresel ve hücre dışı dehidratasyona neden olur ve polidipsiyle sonuçlanan susama hissi uyarılır. Santral diyabetes insipidusun nedenleri nefrojenikten tamamen farklıdır.
Diyabetes insipidus (santral veya nefrojenik) primer polidipsiden, kompulsif bir susama bozukluğuna bağlı olarak hastalar günlük 5- 10 litre kadar sıvı alırlar ve bu durum azalmış ADH salınması ve diüreze yol açar, kesinlikle ayrılmalıdır. Diyabetes insipiduslu hastaların soğuk sıvılar tercih etmesi ayırıcı bir klinik özeııik olabilir. Diyabetes insipidus tanısını onaylamak için ve primer polidipsiden ayırmak için çeşitli testler yapılabilir. İlk olarak sodyum ve osmolariteyi değerlendirmek için rastgele eş zamanlı olarak plazma ve serum örnekleri alınır. Diyabetes insipidusta (santral veya nefrojenik) uygunsuz diürez plazma ozmolaritesinden daha düşük bir idrar ozmolaritesine neden olur. Hastanın hidrasyon durumuna göre plazma osmolaritesi yükselebilir.
Ancak primer polidipside hem plazma hem de idrar ozmolarİtesi dilüe olur. Poliüri nedenlerini ayırmak için kullanılan ana test su kısıtlama testidir. Hastada12-18 ay süreyle sıvı alımı kısıtlanır, ve vücut ağırlığı, kan basıncı, idrar hacmi, idrar dansitesi, ve plazma ve idrar ozmolaritesi her 2 saatte bir ölçülür. Test sırasında dikkatli bir izlem gereklidir çünkü diyabetes insipitusu olan hastalar su kısıtlandığı zaman hızla dehidrate ve hipotansif olabilirler, ve bu durumda test sonlandırılmalıdır. Normalde su kısıtlama sonrası idrar çıkışı 0.5 mL/dk azalır, aynı zamanda idrar konsantrasyonu da plazmadan büyük olur. Diyabetes insipidus (santral ya da nefrojenik) hastalarında su kısıtlamasına rağmen idrar çıkışı yüksek seyreder ve dilüe (israr dansitesi 1005) olmaya devam eder. Primer polidipsi olan hastalarda ise su kısıtlama sonrası idrar ozmolarİtesi plazma ozmolaritesden daha yüksek olur. Su kısıtlamasına idrar ozmolarİtesi plato yapana kadar devam edilir (ardışık 3 saat boyunca saatlik idrar ozmolaritesi artışı 30 mOsm/kg). Bu noktada subkutan olarak 5 ünite vazopressin uygulanır ve idrara ozmolaritesi 1 saat sonra ölçülür. Tam santral diyabetes insipidusu olan hastalarda idrar ozmolaritesi plazma ozmolaritesinin üzerine çıkar, ancak nefrojenik diyabetes insipidusu olanlarda ADH’ya cevap olarak idrar ozmolarİtesi %50’den daha az artar. Kısmi diyabetes insipidusu olan hastalar da idrar ozmolaritesinde artış olur fakat bu artış %50’den azdır, primer polidipsi hastalarında ise bu artış %10dan azadır. Su kısıtlama testi sırasında ADH düzeyleri de ölçülmelidir.
Nefrojenik diyabetes insipidusu olan hastalarda su kısıtlama sırasında ADH düzeyleri normal veya artmıştır, ancak tam diyabetes insipidusu olan hastalarda ise ADH düzeyleri baskılıdır. Kısmi santral diyabetes insipidusu olan hastalarda ise su kısıtlama sırasında plazma ADH düzeyi normalden daha az artış gösterir. Hastalar vazopressin etkisi geçen e kadar (4-8 saat), semptomatik hiponatremi oluşmaması için, fazla miktarda sıvı almamaları yönünde uyarılmalıdırlar.
Santral Diyabetes İnsipidus: Dezmopressin asetat (DDAVP), ADH’nın sentetik analoğu, diyabetes insipidus tedavisinde intranazal veya oral olarak uygulanır. ADH’dan daha düşük pressör: antidiüretik oranına sahiptir. Uygulama frekansı hastalığın ağırlığına göre belirlenir. Tedavinin yeterliliği serum ozmolarite ve sodyum ölçümü ile moniterize edilir. Eğer mümkünse altta yatan altta yatan hastalık durumu düzeltilmelidir. Nefrojenik diyabetes insipidusun spesifik tedavisinde amaçlanan böbreklerdeki solüt yükünü azaltmak için hafif sodyum kaybı sağlamaktır ve bu durumun sonucunda proksimal tübülde geri emilim artar. Ayrıca bu amaca ulaşmak için diüretiklere diyette sodyum kısıtlaması gereklidir.