Hematopoez, vücudun bol çeşitlilikteki hücresel elemanlarının oluşum ve gelişimini belirleyen bir süreçtir. Periferik kan bileşenleri, karmaşık ve dikkatlice düzenlenmiş bir işlem olan hücrenin oluşum sürecinde ortaya çıkar. Pluripotent kök hücre, hem yenilenip hem de değişik nesil hücrelere farklılaşarak dolaşan kandaki uygun tip ve sayıda hücreleri oluşturur. İlkel hücrelerden, özelleşmiş, değişik miktar ve yaşam süreleri olan son dönem hücrelere kadar, olgunlaşmanın döngüsüne sürekli giren hematopoetik sistem, bu özelliği nedeniyle benzersizdir. Kemik iliği, yaşlılık, tüketim ve doku boşluklarına göç nedeniyle hematopoetik hücrelerin döngüsünü karşılayacak kapasiteye sahip olmalıdır. Ayrıca, kanarna, enfeksiyon veya başka stresIere bağlı olarak ortaya çıkan beklenmedik istemlere karşı cevap verecek yedek hücre oluşturma kapasitesi de bulunmalıdır. Hücresel oluşum ve dönüşümün tekrarlayan döngülerini anlamak, hematolojinin normal ve patolojik mekanizmaları hakkında önemli görüşler sağlayabilir.
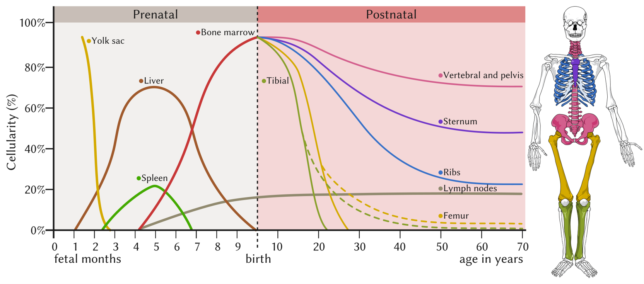
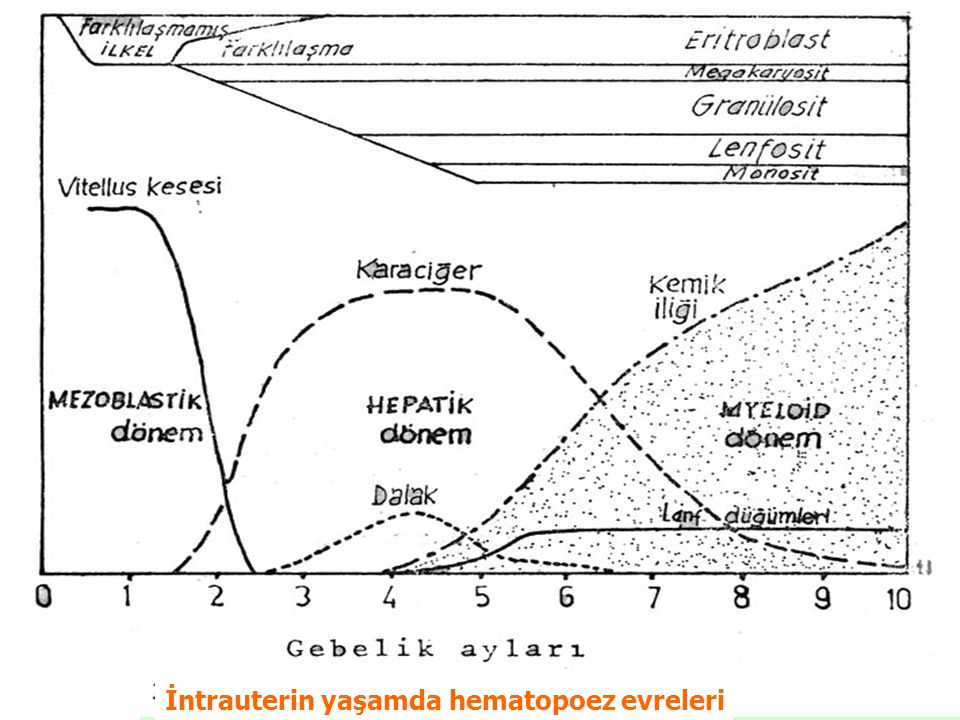
Hematopoez, ilk hemoglobinize hücrelerin erken eritroblastlarının adacıklar oluşturduğu, embronik yolk salkta başlar. 6 haftalık gestasyondan sonra fetal karaciğer, ilkel lenfositoid hücreler, megakaryositler ve eritroblastları oluşturmaya başlar ve dalak ikincil bir eritropoez bölgesi haline gelir. Hematopoez sonradan kemik iliğine geçer. Erken yaşamda, tüm fetal kemikler bu rejeneratif kemik iliğini içerirler, ancak yaşla birlikte bu kemik ilikleri yağla dolar. Erişkinde, aktif kemik iliği sadece aksiyel omurga (sternum, vertebralar, pelvis ve kaburgalar) ile femur ve hUlnerusun proksimal sonlarında bulunur. Bu nedenle, birçok hematolojik tanı için gereken kemik iliği örnekleri iliak kanat veya sternumdan alınır. Kemik iliği fibrozisiyle giden hastalıklar (miyeloproliferatif hastalıklar) veya ciddi kalıtsal hemolitik anemiler (talasemi major) gibi kemik iliği boşluğunu baskılayan patolojik durumlarda, ekstramedüller hematopoez, başta dalak olmak üzere fötal hematopoez bölgelerinde yeniden başlayabilir.
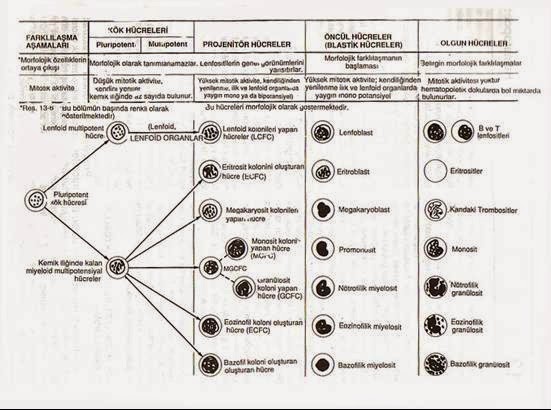
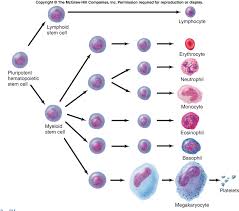
Tüm olgun hematopoetik hücrelerin küçük bir pluripotent kök hücre topluluğundan köken aldığı kabul edilir. Kemik iliğindeki hücrelerin % 1’ini oluşturan bu hücreler, hiçbir yapısal işaret taşımazlar ve en iyi şekilde, benzersiz işlev özellikleriyle tanımlanırlar. Kök hücreler iki özellikleriyle ayrılır. Öncelikle oldukça esnek ve üretkendirIer, büyük sayılarda granülosit, lenfosit ve eritrositleri hayat boyunca ikmal ederler. Sürekli olarak kan hücreleri sağlama gereksinimi, seçilmiş hücreleri kısa bir sürede büyük miktarlarda üretme kapasitesine gereksinim duyar. Örneğin, mikroorganizmaların saldırısıyla oluşan bir enfeksiyon nötrofillerin salınımını tetiklerken, hipoksi veya akut kan kaybı kırmızı küre üretimini arttırır. İkinci olarak da, kök hücreler kendi kendilerini yenileyerek değişik nesil hücrelerin ön hücrelerini devamlı olarak sağlarlar. Bu büyük üretim potansiyeline rağmen normal durumlarda pasiftirler ve sadece çok az hücre artar veya farklılaşır. Yine de üretme yetenekleri göze çarpar. Ölümcül radyasyona maruz kalan farelerle yapılan çalışmalar çok az nakledilen hücrenin (kololü oluşturan ünite dalak hücreleri- CFU-S) bile, çoklu nesil oluşturan hematopoez yeteneklerini göstermiştir.
Pluripotent kök hücrelerin ön hücrelere farklılaşmasını düzenleyen sinyaller bilinmemektedir. Veriler, nesil oluşumuna giden ilk adımın stokastik (şans) olayı olduğunu düşündürmektedir; olgunlaşmanın sonraki evrelerinin büyüme faktörleri veya sitokinlerin etkisi altında oluştuğu varsayılır. Sitokinler, özgül sitokin reseptörleri aracılığıyla değişik hücreler üzerine etkir. Bu reseptörlerin aktivasyonu, gen transkripsiyonunda değişiklikler yapan ve sonuçta hücre proliferasyonu ve farklılaşmasına yol açan sinyal transdüksiyon yolaklarını indükler. Bu büyüme faktörlerinin, aynı zamanda apoptozisi (programlanmış hücre ölümü) önleyerek, hematopoetik hücrelerin gelişiminde hayati etkileri olduğu gösterilmiştir. Bu süreç, kemik iliğinin hücresel çevresinde oluşur. İyi hatırlanması gereken bir başka nokta da, hematoiezisin hematopoetik olmayan hücrelere de bağlı olduğu (fibroblastlar, endotelial hücreler, osteoblastlar ve yağ hücreleri) ve bunların kemik iliği mikro çevresini oluşturduğu kadarıdır. Kök hücre biyolojisi, bölgeselolarak ve kök hücrelerle bunları çevreleyen parankim arasındaki hücre yüzey bağ etkileşimleriyle üretilen hematopoetik sitokinler tarafından düzenlenir.
Geleneksel olarak, hematopoezin, kemik iliğindeki sitokinler ve intrensek transkripsiyon faktörlerinin ayarladığı sıkı bir hiyerarşi içinde düzenlendiği varsayılır. Daha ilkel hücrelerin özgül düzenleyici sitokinlerin etkisi altında olgunlaşması nedeniyle, birçok hücre bölünmeye uğrar ve bir soya ait öncül hücreler haline gelir. Kendini yenileme kapasitelerini de kaybederler. Morfolojik olarak bu hücreler, özgül olmayan blasta benzer hücrelerden renk, şekil, granüler ve çekirdek içerikleriyle ayırt edilebilen hücrelere dönüşürler. İşlevsel olarak, ayırt edici hücre yüzey reseptörleri ve özgül sinyallere yanıt kazanırlar. Olgunlaşan granülosit ve eritroid hücreler, kemik iliğinde birçok bölünmeye uğrarken lenfositler, timusa ve lenf düğümlerine olgunlaşmak için göç ederler. Megakaryositler, bölünmeye son verir ve çekirdek replikasyonu yaparlar. Sonunda bu hücreler kemik iliğinden, tümüyle işlevsel durumdaki eritrosit, mast hücreleri, granülosit, monosit, eozinofil, makrofaj ve trombositler olarak salınırlar.
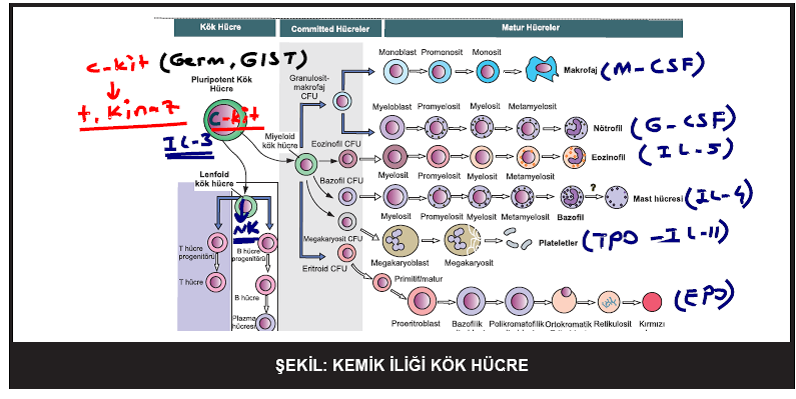
Pluripotent kök hücre, morfolojik olarak ayırt edilemez ve en iyi şekilde, hücre farklılaşma antijeni CD4 ve in vitro olarak pluripotent koloniler oluşturabilmesiyle tanımlanabilir. Interlökinler (IL) -I, -3 ve -6, fms benzeritirozin kinaz 3 (flt 3) ve özgül bir kök hücre faktörü (c-kit bağı, veya steel faktörün etkisi altında, bu hücre, ya miyeloid seri kök hücre (CFU-granülosit-eritrositmakrofaj- megakaryosit [CFU-GEMM] veya lenfoid seri kök hücreye olgunlaşabilir. İlkel eritroid öncüller, miyeloid kök hücreden köken alan, çoğalma oluşturan (burst-forming) eritroid birim hücrelerdir. Bu hücreler daha sonra, CFU-eritroid (CFU-E) hücrelere farklılaşır ki bunlar eritrositlerin öncülleridir. CFU-E hücreler, eritropoetin (EPO) için reseptörler içerir. Bu, 18-kd bir molekuldür ve düşük oksijen durumları veya anemiye yanıt olarak böbrek interstisyel hücreleri tarafından üretilir. EPO, CFU-E hücrelerinin çoğalmasını düzenler ve hemoglobin sentezleyecek olan proeritroblast ve retikulositlere olgunlaşmalarını destekler.
İnsan GM-CSF maddesi, hematopoetik yolakta CFUGEMM kök hücrenin olgunlaşmasını düzenleyecek şekilde, erken evrede rol alır. Miyeloid öncüllerin farklılaşması, granulosit-CSF (G-CSF) ve monosit CSF’nin (M-CSF) yönlendirmesiyle olur. CFU granulosit hücreler, birçok segmente ayrılan çekirdekleriyle kolayca hatırlanan miyeloblast, miyelosit ve erken polimorfonükleer nötrofiilere dönüşürler. CFU-monosit hücreler ise, monoblastlardan promonosit ve monositlere, bazen de makrofajlara dönüşürken hep tek hücreli olarak kalır.
Eozinofil ve bazofiller, CFU-GEMM hücrelerden, sırasıyla IL-S ve IL-3–IL-4 etkisiyle oluşur. Özgül granüler içerikleri, öncül hücreleri erken monositIerden ayırmamıza yardımcı olur. Trombosit gelişimi morfolojik olarak diğer seri lerden farklıdır. CFU-GMM, CFU-megakaryosite dönüşür. Bu şekilde adlandırılmasının nedeni, erkenden hücre bölünmesine uğradıkları halde, çekirdeklerinin replike olmamasındandır. Megakaryositler, vücutta DNA içeriğini ikiye katlayabilen tek hücre grubudur (endomitoz). Birçok hücresel döngüden sonra olgunlaşan megakaryosit, trombositlere dönüşür. İki büyüme faktörü, trombopoetin ve IL J J’in hem hayvan, hem de İnsan deneylerinde trombosit sayısını arttırdığı, bunu da özellikle megakaryosit gelişimini arttırarak yaptığı gösterilmiştir.
Son bulgular, hematopoetik kök farklılaşma hiyerarşisinin karışıklığını ortaya koymaktadır. Hematopoetik kök hücreler, hem immatür öncüller, hem de çapraz serilerden non lenfo hematopoetik hücreler, miyozit, hepatosit, gastrointestinal epitelyal hücreler ve nöronlar gibi hücrelere farklılaşır. Bu plastisitenin nedeninin, yetişkin hematopoetik sisteminin intrensek mi, yoksa eksvivo olarak hematopoetik hücrelerin diğer dokularla birleşmesi sonucu mu olduğu tam açık değildir. Yine de, yetişkin hematopoetik kök hücrelerin dinamik yeniJenebilen bir doku onarım ve rejenerasyon kaynağı olduğunu varsaymak, gelecek için umut vaat etmektedir.
Normal hematopoezi etkileyen faktörlerin bulunuşuyla, hematopoetik hücre oluşumunda defekti olan hastaların tedavisinde önemli gelişme kaydedilmiştir. Özgül sitokinlerin varlığında, her serinin hematopoetik hücreleri, çoğalmak ve farklılaşmak üzere stimule edilebilmektedir. DNA teknolojisindeki yenilikler, rekombinan insan (rh) proteinlerinin, in vivo aktivitesine benzer şekilde sentezi ve pürifikasyonuna olanak vermektedir. Hastalara bu ürünlerin uygulanması, periferik kana olgun hücrelerin salınmasını sağlar. Örneğin eksojen EPO, böbrek yetmezliği, kemoterapi ve ilik yetmezliği sendromlannda oluşan aneminin tedavisinde kullanılması değerlendirilmektedir. Kanserli hastaların kemoterapileri sonrasında rhIL-ll uygulanması, trombositopeni görülme insidansında azalma sağlamaktadır. Kemoterapi veya radyoterapi sonrası nötropeni gelişen hastalarda rhG-CSF uygulamasının, hastane kalışıarında ve yüksek enfeksiyon risk süresinde kısalmaya neden olduğu gösterilmiştir. Sitokinlerin ayrıca, gecikmiş kök hücre aşılaması olan hastalarda, kök hücre naklinden önce ve sonra yapılan koleksiyonlardan periferal kök hücrelerin mobilizasyonundada rolleri mevcuttur.
Hematopoetik kök hücre biyolojisinin anlaşılmasındaki artış, kök hücrelerin tedavi amacıyla manipule edilme tekniklerinin gelişimini arttırmıştır. çoğu antikemoterapötik ilaçlar ve radyasyon tedavisinin antitümör etkileri doza bağlıdır ve her ikisinin de en önemli doz kısıtlayan toksisitesinin miyelosupresyon olduğu uzun zamandır bilinmektedir. Hematopoetik kök hücre nakli, kemoterapinin yoğun miyeloblastif dozlarının uygulanmasına olanak tanır ve tüm vücut radyasyonu, malign hücre eradikasyonunu sağlar. Bunu, abi ase kemik iliğini doldurmak için kök hücrelerinin infüzyonu (donor veya aynı hastadan olabilir) takip eder. Transplantasyonun tedavi edici potansiyeli, daha önceleri lösemi gibi birincil kök hücre bozukluklarında kullanılırken, artık malign olmayan hematolojik hastalıklarda (örneğin, aplastik anemi, orak hücreli anemi, doğumsal immün yetmezlikler), solid tümörler (örneğin, renal hücreli karsinom, melanom) ve malign olmayan otoimmün hastalıklarda (örneğin, amiloidoz, sistemik lupus) değerlendirilmektedir. Genellikle daha genç hastalar «50 yaş), bu yoğun tedavi için en iyi adaydır. Kök hücre naklinin birçok yolu geliştirilmiştir. Otolog nakilde, hastanın kemik iliği veya periferik kandaki kök hücreler, yüksek doz kemoterapiyi ve/veya rhG-CSF uygulamasını takiben oluşan remisyon sırasında toplanır. Bu hücreler dondurulur ve çözdürülerek reinfüze edilirler. Böyle bir yaklaşımla, tümörle kontamine olabilen kök hücre ürününün reinfüzyonu sonucu daha yüksek bir relaps riski oluşur. Allojenik nakil ise, anormal işlev gören kemik iliği eradike edildiğinde ve/veya akraba olsun olmasın uygun bir kaynaktan gelen kök hücrede uygulanan bir işlemdir. Tüm vücut radyasonuyla birlikte veya tek olarak uygulanan yüksek doz kemoterapi, hastanın kemik iliğin i yıkmak için kullanılır ve sonrasında normal hematopoezi aşılayan ve restore eden yeni kök hücrelerin infuzyonu yapılır. Bu nakilde, tedavi bağımlı morbidite önemlidir, işlem bağımlı mortalite de %10-30 oranında görülür. Ancak, nakledilen ilikteki intakt lenfositlerin hastanın kendi dokularına yaptığı saldırı sonucu oluşan ve bir otoimmün fenomen olan, Altcıya-Karşı-Doku-Hastaltğı’nı (GVHD) baskı lamak için yapılan destekleyici ve immunmodulator tedavisi geliştirilmesi, sonucu iyileştirmektedir. Verici ve hasta, tüm hücrelerdeki insan lökosit antijeni (HLA) ve major histokompatibilite kompleks (MHC) proteinleri açısından teste tabi tutulmalıdır.
Üç major HLA Sınıf i antijen (A, B ve c) ve üç MHC Sınıf II antijen (DP, DQ ve DR) geliştirilmiştir. Altı HLA gen loküsü, 6. kromozama sıkıca bağlıdır ve bunlar, gen veya haplotipler kümesi şeklinde miras alınmaktadır. Böylece tüm çocuklar, her bir ebeveyni ile yarı eş durumdadır (haploidentikal) ve öz kardeşler birbirlerinin %25 oranında HLA eştir. HLA uygunluğu olan, ancak akraba olmayan nakillerde HLA uygun akraba olanlara göre, diğer minor HLA doku uygunsuzluklarına bağlı olarak, daha fazla GVHD görülmektedir. HLA uygunluğu olmayan kök hücreleri alan hastalar akut GVHD, ilik reddi ve ölümcül ilik aplazisi riski taşır. HLA uyumu olmayan nakilli hastalardaki (6 veya 5’ten az HLA loküsü eşleşen), mortalite ve morbidite önlenebilir. Singeneik nakil, ikizinden kemik iliği alırsa söz konusu olur. Bu vakalarda, hasta ve donör ideal olarak eşleşmiştir. Her ne kadar primer hastalığa karşı immun yanıt yokluğundan dolayı relaps sıklığı artsa da (graft versus lösemi, sonra anlatılacaktır), uzun dönem sonuçlar çok iyidir. Daha önceleri nakillerde, vericinin posterior iliak kanadından aspire edilen ve myeloablasyon ya da immunsupresif tedavi sonrası hastaya intravenöz olarak infuze edilen allogenik kemik iliği hematopoetik kök hücreleri kullanılmaktaydı.
Normal hematopoetik işlevin aşılama veya yeniden yapılanması süreci haftalar alabilir. Bu uzamış nötropeni döneminde genelde, hastalar günlük trombosit ve kırmızı hücre transfuzyonuna ihtiyaç duyar ve hayatı tehdit eden bakteriyel, viral ve fungal enfeksiyon riskini en aza indirmek için hastaneye yatırılır. Diğer komplikasyonlar, ciddi mukozit, hemorajik sistit, GVHD, tekrarlayan hastalık ve doku yetmezliğidir. Kök hücre naklindeki yeni teknolojiler, yüksek doz rhG-CSF tedavisinin, kemik iliğinden büyük miktarlarda CD34+ hematopoetik progenitor ve kök hücrenin dolaşan kana salıverilmesini sağladığının keşfiyle, daha da güçlenmiştir. Birçok çalışmada, yüksek doz fhG-CSF tedavisi olan sağlıklı bireylerde, dolaşımdaki CD34+ konsantrasyonunda geçici olarak bazal düzeyin 10-15 katı artış olduğu gösterilmiştir. Bu mobilize hematopoetikhücreler, daha sonra aferezis işlemleriyle toplanır ve nakil için kemik iliği hücrelerinin içine konur. Kemik iliğinden alınan kök hücrelerle karşılaştırıldığında, periferik kandan alınanlar, daha kolayaşılanır ve nötrofil, eritrosit ve trombositlerin miyeloablasyon sonrası daha iyi iyileşmesini sağlar. Primer hematolojik hastalıkları nedeniyle allogenik periferal kök hücre nakli yapılan hastalar, nötrofil iyileşmesi için daha kısa zaman, dahaaz transfuzyon, akut GVHD için daha az hastanede kalış süresi ve kemik iliği nakli olan hastalara benzer uzun dönem sağkalım gösterir. Periferal kan kök hücre koleksiyonları, saklanmış ilik dokularına göre, 3-4 kat fazla CD34+ ve LO fazla lenfoid hücre içerdiğinden, kronik GVHD görülme sıklığında artış olabilir. Umblikal kord kanının bir diğer zengin CD34+ kaynağı olduğunun keşfedilmesiyle, başarılı kord kanı kök hücre nakilleri yapılmıştır. Bazı çalışmalar, geleneksel ilik ve periferal kan nakillerine benzer uzun dönem sonuçlara sahip olduğunu göstermiştir. HLA eşlemeli vericisi olmayan hastalar için bir tedavi seçeneği olsa da, saklanmış kordlarda daha düşük CD34+ hücrelerinin bulunması, bu işlemi çocuklar ve küçük yapılı hastalarla sınırlandırılmıştır. Giderek artan miktarda kanıt, bazı hastalarda hematopoetik kök hücre nakline bu kadar iyi cevap görülmesinin, kısmen, yeni nakledilmiş dokunun hastalığı aktif baskılanmasına bağlı olduğunu göstermektedir ve lösemiye karşı doku olarak adlandırılmaktadır.
Kronik miyelogenöz lösemi (KML) için allogenik nakil yapılmış ve relaps kamtı olan hastalarda, verici lenfositlerinin infuzyonunun remisyonu sağladığı çalışmalarla gösterilmiştir. Buna karşıt olarak, verici ve alıcı arasındaki etkileşimi en aza indirmeye çalışan işlemler, hastalık relapsını arttırmaktadır. Örneğin, singeneik nakil ve GVHD’i azaltmak için T hücresi alınmış ilik nakli yapılmış hastalarda relaps görülme olasılığı artmıştır. Verici lenfosit infuzyonlarının KML’yi kontrol edici etkinliğinin gözlenmesi, nakledilmiş allogenik hücrelerin immunolojik etkilerinin, bazı hematolojik malinitelerin tedavisi için sitoredüksiyon kadar (veya daha fazla) önemli olabileceği sonucunu vermiştir. Bu etkileriyle ayrıca, agresif sitoredüksiyon olmadan verici kök hücre aşılamasına olanak verecek dozlarda, düzenleyici ve immunsüprese tedavi alan hastalarda nonmyeloablatif nakiller yapılmaktadır. Bu mini-transplantlar, sitopeni veya hematopoetik kompromize dönemleri olmadan, kimerik kemik iliğiyle sonuçlanır (yarı alıcı yarı verici). Zamanla, yanıt veren hastaların büyük çoğunluğunda kemik iliği tamamen vericininkine döner. Hala deneysel olmasına rağmen bu işlemler, geleneksel myeloablatif nakil rejimIerine uygun olmayan 50 yaş üstü ve/veya komorbiditeleri olan, ya da malign olmayan otoimmun bozuklukları olan hastalarda giderek artan oranlarda kullanılmaktadır.
Hematopoetik kök hücre hastalıkları, kök hücre gelişiminin normal düzenini bozar ve olgun ürünlerin az yapımı (aplastik anemi), fazla yapımı (miyeloproliferatif hastalıklar) veya olgun olmayan formların aşırı yapımıyla karakterize olan bozulmuş farklılaşmayla (miyelodisplazi ve akut lösemi) sonuçlanır. Miyeloproliferatif, miyelodisplastik ve lösemik bozukluklar tartışılmıştır. Hematopoetik kök hücre yetmezliği aplastik anemiye (AA) yol açar. Bu, pansitopeni (kan hücre serilerinin azalmış üretimi) ve belirgin hiposellüler kemik iliğiyle karakterize nadir bir bozukluktur. ilk olarak 1888’de Paul Erhlich tarafından, genç bir kadın hastanın otopsisi sırasında, ciddi anemi ve ileri derecede hipoplastik nötropenili kemik iliği üzerinde, tanımlanmıştır. Daha yeni çalışmalar, ciddi AA olan hastaların normal fonksiyonel ilik stromal hücreler ile, normal hatta yükselmiş uyarıcı sitokinleri olmasına rağmen, pluripotent kök hücrelerinin sadece bir parçasının normalolduğunu göstermiştir. AA yaygın değildir. Genel populasyondaki insidansı milyonda 1-5’tir. Geneııikle 20-25 yaş civarı genç yetişkinlerde ve 60-65 yaş arası yaşlı yetişkinlerde görülür. Gelişmekte olan ülkelerde (Tayland ve Çin), sanayileşmiş toplumlara göre (Avrupa ve israil) üç kat fazla görülür. Bu fark, radyasyon veya ilaçla açıklanamaz. AA vakalarının küçük bir oranı, doğumsal kemik iliği yetmezliği olarak kendini gösterir, Fanconi anemisi, Schwachmann Diamond sendromu ve diskeratosis konjenita gibi. En yaygın olanı Fanconi anemisidir. Bu, DNA tamiri yapan proteinleri kodlayan genlerdeki mutasyonlardan kaynaklanan otozomal-resesif bir bozukluktur. Otozomal dominant otan diskeratosis konjenita, telomeraz kompleksleri için olan genlerdeki mutasyonlar sonucu olur. ilik yetmezliği ve prematür yaşlanmayla karakterizedir. Bilinen AA nedenleri çok çeşitlidir ve miyeloablatif radyasyona maruziyetten, yaygın virüs ve ilaçlara kadar değişir. ilaç, kimyasaııar (benzen, petrol ürünlerindeki siklik hidrokarbonlar, lastik yapıştırıcı, insektisidler, kimyasal boyalar) veya radyasyona önceden maruz kalmış olmak hastada AA oluşumuna zemin hazırlar, çünkü bu maddeler DNA harabiyetini arttırarak doğrudan kök hücre çoğalması ve farklılaşmasını bozar. Buna zıt olarak, sitotoksik kemoterapiler (özellikle alkilleyici ajanlarla yapılan) ve radyasyon gibi tedaviler, bütün hızlı döngüsü olan hücreleri hedef alır ve genellikle geriye dönebilir kemik iliği apıazisine neden olur. Edinilmiş AA’nin birçok nedenine rağmen, genelde vakalarınçoğu idiyopatiktir. Şu ana kadar ki veriler, otoreaktif alıcı lenfositlerinin, AA’daki normal hematopoezin harabiyetinin asıl sorumlusu olduğunu göstermektedir. Kemik iliği stromal hücreleri ve sitokin düzeyleri, AA’1i hastalarda normaldir. AA’nin immün düzenleme bozukluğu hastalıklarında ve viral enfeksiyonlardan sonra görülmesi, hastalık mekanizmasının immun aracılı olduğunu düşündürmektedir. Bir hipotez, viruslerin veya ilaçların, immün sisteme verilen antijenterin, sitotoksik T hücre yanıtlarını tetiklediği ve normal kök hücreleri harap ettiğini savunur.
Nadir vakalarda, yüzbinde bir hasta, ciddi bir idiyosenkratik ilaç reaksiyonu geliştirir. Ancak bu hastaların maruziyete (non-steroid antiinflamatuar ilaçlar, sülfonamidler veya Ebstein-Barr virus gibi) duyarlı bir genetik predispozisyonu olup olmadığı bilinmemektedir. AA’nın klinik başlangıcı sinsi ve ani olabilir. Hastalar genellikle sitopeniyle ilişkili belirtilerden şikayet ederler: anemi nedeniyle güçsüzlük, yorgunluk, dispne veya çarpıntı; düşük trombositlere bağlı dişeti kanarnaları, epistaksis, peteşi veya purpura; düşük sayıdaki veya işlev görmeyen nötrofillere bağlı rekürren bakteriyel enfeksiyonlar. Fizik muayene, çok sayıda anormalliğe sahip olabilen konjenital AA’li hastalar dışında, genellikle normaldir.
Pansitopeninin ayırıcı tanısı geniştir ve birincil kemik iliği bozuklukları (AA, hiposellüler miyelodisplastik sendrom (MDS); akut lösemi ve paroksismal noktürnal hemoglobinüri) ile kemik iliğini tutan sistemik hastalıklar olarak ayrılır. AA’nın tanısal doğrulaması, kemik iliği biyopsisinde hiposellülaritenin kanıtlanması ve diğer ilik proseslerinin ekarte edilmesiyle mümkündür. Normal kemik iliği sellülaritesi, 70 yaşa kadar %30-50 ve 70 yaş üstünde %20’nin altındadır. Tersine, AA’li hastalarda sellülarite %5-15 arasında olup, artmış yağ depolanması ile, varsa çok az hematopoetik hücreler görülür (öncelikle plazma hücreleri ve lenfositler). AA’de hematopoetik progenitor ve öncü (prekürsör) hücreler morfolojik olarak normaldir fakat, normal hücrelerin %1’inden azdır ve in vitro olarak, farklılaşmış öncül hücre kolonilerini oluşturma yeteneğinin azalarak işlevsizleştiği gösterilmiştir. Blastların artması, displastik hematopoetik hücrelerin (pseudo-Pelger-Huet anormallikleri veya mikromegakaryositler gibi) ( ya da, periferik kan veya ilikteki klonal sitoıgenetik anormal hücrelerin varlığı, hiposellüler bir ilik olsa bile, AA’yi değil, akut lösemi veya MDS’yi düşündürür. Genç hastalarda Fanconi anemisi tanısı, kültüre hücrelerin, mitomisin veya diepoksibütanla indüklenen kromozomal harabiyete olan artmış duyarlılığının gösterilmesiyle yapılır. Her ne kadar, AA’li hastalar periferik yaymada düşük retikülosit sayımıyla (düşük kırım hücre üretimine bağlı) birlikte kan hücrelerinin azlığı ve makrositik hücreler gösterse de, başka birincililik bozuklukları olan hastalarda da benzer bulgular görülebilir.
AA’nin tedavisi hastalığın ciddiyetine dayalıdır. Hafif sitopenili hastalar izlenebilir. Halbuki, periferik kan sayımına dayalı ciddi AA’li hastalar tedavi edilmedikleri takdirde, 2-6 ayarasında değişen sağkalım oranlarına sahiptir. Birçok hasta kaçınılmaz enfeksiyonlardan öldüğü için, ileri derecede nötropeni li hastalarda geniş spektrumlu antibiyotikler, antifungal ve antiviral ajanlarla destekleyici bakım sağlanmalıdır. Kırmızı hücre ve trombosit transfuzyonları, ciddi belirtileri olan hastalarda yardımcı olabilir (nakil için uygun hastalara bakımla birlikte). AA tedavisinde güncel yaklaşımlar, defektif kök hücrelerin nakille yerine koyma veya aşırı aktif immün yanıtın kontrolüne odaklanmıştır. Ciddi AA olan genç hastalar ve HLA uygun iliği olan vericiler, allogenik ilik nakli için değerlendirilmelidir. Bu işlem normal kök hücre işlevini yerine koymayı amaçlar ve hastalık için en iyi tedaviyi sunar. Her ne kadar, kardeş vericiden nakil yapılmış 30 yaş altı hastalarda uzun dönem sağkalımı mükemmel olsa da (%75-90), transplantın kendisinden kaynaklanan morbidite ve nakille ilişkili uzun dönem komplikasyonlarının yönetimi hala süren sorunlardır. 40 yaş üstü veya HLA uyumu olmayan vericiden nakil yapılmış hastalarda sonuçlar kötüdür. ilaçların indüklediği apıazinin varsayılan immün mekanizması, yaşlı hastalardaki AA, uygun kök hücreverici si bulamayan hastalar ve kök hücre nakline uygun olmayan hastalar için immünsupresif tedavi yaklaşımlarının düşünülmesini sağlamıştır.

Antitimosit globulin (ATO) ve siklosporin (özgül bir T hücre baskılayıcısı) kombinasyon tedavisi, %70-80 hastada ilik işlevlerinin (örneğin, kırmızı hücre veya trombosit transfuzyonuna gerek duymamak) geriye gelmesini ve yanıt verenlerde 5 yıllık %90 sağkalım oranı sağlar. ATO ve ALG’nin yan etkileri içinde yer alan anaflaksi, serum hastalığı (antiserumdaki at veya tavşan antijenlerinin sonucu) genelde kendini sınırlar. Bu hastalıkta genellikle relaps ve rekürrensler olur ve tekrarlayan ATO, yeni immunsüpresif ajanlar (mikofenolat mofetil gibi), androjenler ve deneysel ajanlarla tedavi gerektirir. Siklofosfamid gibi geleneksel kemoterapilerin oldukça toksik olduğu kanıtlanmıştır. AA’li hastalarda endojen sitokinlerin yüksek olması nedeniyle, rhO-CSP, EPO veya kök hücre faktörü gibi büyüme faktörlerinin rutin kullanımı genellikle etkisizdir. Yine de, dirençli hastalarda, sitokin kombinasyonunun uzun süreli uygulanmasının, kan hücre sayımının devamı için bazı etkileri mevcuttur. ilk aplazi tedavisinde hayatta kalan hastaların, miyelodisplazi, lösemi ve paroksismal noktürnal hemoglobinüri gibi diğer birincil hematolojik bozukluk riski vardır. Bu tür klonal hastalıklarla orijinal apıastik anemİ patogenezi arasındaki ilişki çelişkilidir. Örneğin, bazı çalışmalarda AA’li hastalarda aynı zamanda PNH klonları bulunduğunun gösterilmesi, bu hastalıkların özellikleri arasında anlamlı bir örtüşme olduğunu düşündürmektedir.