Eritrositler (RBC), bedendeki tüm dokulara oksijen sağlar, karbon dioksiti atılmak üzere akciğerlere taşırlar. Bu işlevler için özelleşen eritrositlerin. İki yönlü konkav disk şekilleri sayesinde gaz alışverişi için yüzey alanları en üst düzeye çıkar ve kılcal damar ağından yeterince geçebi lecek şekilde uyum sağlayacakları bir zar yapısı ve hücre iskeletine sahiptirler. Zar proteinleri ( band 3 ve gliforin) ve iskelet yapısını oluşturan sitoplazmik proteinler (spektrin, ankrin ve protein 4. 1) arasındaki etkileşimler sayesinde eritrositlerin, dinlenme halindeki çaplarının dörtte biri kadar olan kılcal damarlar arasından geçişi mümkün olabilmektedir. Olgun eritrositin çekirdeği yoktur ve yaşam süresi, kemik iliğinden dolaşıma salıverilmesinden ve çekirdeğin atılmasından önce sentezlenen proteinlere bağlıdır. Olgun eritrositin sitoplazmik proteininin yaklaşık %98’ini hemoglobin oluşturur. Geri kalanı, anaerobik metabolizma ve heksoz monofosfat şantı için gereken proteinler gibi başlıca enzimatik proteinlerdir. Bir sonraki bölümde de anlatıldığı gibi, eritrositin iç yapı özelliklerindeki herhangi bir hata, hemolitik anemiyle sonuçlanabilir. Zar ve hücre iskeleti proteinlerindeki anormallikler, eritrosit şekil ve esnekliğinin değişmesine neden olurlar. Glukoz metabolizmasında yer alan enzimatik yollardaki doğumsal defektier, oksidatif strese direnci azaltır, hemoglobin yapı ve sentezindeki kalıtsal anormallikler, anormal hemoglobinin (orak hücre hastalığı) polimerizasyonuna veya dengesiz hemoglobin zincirinin (talasemi) çökmesine yol açar. Tüm bu değişiklikler eritrosit ömrünün azalmasıyla sonuçlanır. Oksijen, 2a ve 2a benzeri zincirden oluşan bir tetramer olan hemoglobinle taşınır. Fötal dönemde bulunan temel hemoglobin, fötal hemoglobindir (HbF)’ HbP’in erişkin hemoglobine HbA (aı-ı) dönüşü, perinatal dönemde gerçekleşir hayatın ilk 4-6 ayına kadar HbF’in total hemoglobine oranı yüzde 1’e kadar düşer. HbAı (aı,Yı), HbA’nın yaklaşık yüzde 1’ini oluşturan minör bir erişkin hemoglobinidir.
Anemi, eritrosit kütlesinin azalmasıdır ve hastalıkların önemli bir belirtisidir. Primer bir hematolojik hastalık nedeniyle ya da sistemik bir hastalığa bağlı eritrosit yapımiilin azaldığını gösterebilir. Ya da tam aksine, hemolize bağlı artmış bir hücre döngüsünün göstergesi olabilir. Bu durum, immün eritrosit harabiyetine primer olarak veya sistemik vasküler bir sürecin bir parçası olarak intrensek eritrosit anormalliklerinden kaynaklanabilir. Aneminin araştırılması, hastanın değerlendirilmesinde önemli bir komponenttir ve genellikle sistemjk bir hastalığın önemli bir işaretidir.48-1 'de aneminin ayırıcı tanısı özetlenmektedir.
Anemi belirtileri genellikle eritrosit kütlesindeki azalmanın hızına bağlıdır. Akut hemoraji veya masif hemolizi olan hastalarda, hipovolemik şok belirtileri görülebilir. Ancak, çoğu hastada anemi, daha yavaş seyreder ve çok az belirtiye neden olabilir. Genel belirtiler, yorgunluk, egzersiz tolerans düşüklüğü, dispne ve palpitasyondur. Koroner arter hastalığı olan hastalarda anemi, göğüs ağrısı belirtilerinin daha da kötüleşmesine yol açabilir. Fizik muayenede aneminin başlıca bulgusu, solukluktur. Hastada taşikardik olabilir ve genellikle dinlemekle duyuiabilen üfürüm bulunabilir. Hemolizli hastalarda ise sıklıkla sarılık ve splenomegali görülür.
Anemide, laboratuvar incelemelerinin anahtar bileşenleri, retikülosit sayımı, periferik yayma, eritrosit indeksleri, beslenmenin değerlendirilmesi, kemik iliği aspirasyon ve biyopsisinden oluşur. Retikülosit sayısının ölçülmesi, primer eritrosit yapım yetersizliğine bağlı anemi ile eritrosit yıkımındaki artıştan kaynaklanan anemilerin ayırımının yapılmasını sağlar. Kemik iliğinden yeni salınmış eritrositler hala küçük miktarlarda RNA’ya sahiptir ve retikü/osit olarak adlandınlırlar ve periferik kan yaymasının metilen mavisi veya diğer supravital boyalarla boyanmasıyla görülebilir. Anemi stresine yanıt olarak eritropoetin (EPO) üretimi artar ve retikülosit yapımı hızlanır veya daha fazla sayıda retikülosit dolaşıma verilir. Bu nedenle, periferik kandaki retikülosit sayısı, kemik iliğinin anemjye verdiği yanıtın göstergesidir. Bu sayı, retikülositin toplam eritrosit sayısına oranı veya mutlak değer şeklinde belirti lebilmektedir. Anemisi olmayan hastalarda normal retikülosİt sayısı %1 ve 50000/mcl’dir. Anemi, eritrosit yaşam süresindeki azalma nedeniyle oluştuğunda, uygun kemik iliği cevabı, % 2’nin üzerinde retikülosit sayısı ve/veya 100000/mcl’inüzerinde mutlak retikülosit sayısıyla sonuçlanır. Retikülosit sayısı artmamış ise, eritrosit yapım eksikliğinin nedeni araştırılmalıdır. Anemide, dolaşımdaki hücrelerin azalmasıyla, kemik iliğinden salınımında herhangi bir artış olmaksızın retikülosit sayısı artacağından, toplam eritrosit yüzdesiyle ifade edilen retikülosit sayısının düzeltilmesi gerekir. Düzeltilmiş retikülosit sayısı, retikülosit sayısının, hastanın hematokritinin normal bir hematokritle çarpılmasıyla hesaplanır. Mutlak retikülosit sayısının avantajı ise, bu düzeltmenin yapılmasını gerektirmemesidir. Mutlak retikülosit sayısı, giderek daha sıklıkla ölçülebilmektedir ve muhtemelen standart retikülosit tayininin yerini alacaktır. Periferik yaymanın değerlendirilmesi, aneminin nedenlerinin tespitinde önemli ipuçları verebilir. Retikülositozun eşlik ettiği aneminin değerlendirilmesinde, eritrosit morfolojisinin incelenmesi, özellikle önemlidir. Periferik yayma ayrıca, immün hemoliz (sferositler ile karakterizedir) ve mikroanjiopatik hemolizin (şistozit veya eritrositlerin parçalanmasına neden olur) ayrımında gereklidir.
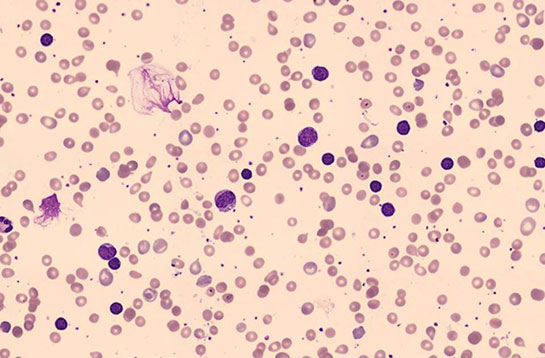
Aneminin diğer nedenlerine eşlik eden değişiklikler, hemoglobinopatilerin karakteristiği olan orak ve hedef hücreleri, myelofibroz ve kemik iliği infiltrasyonuyla ilişkiligözyaşı hücrelerini ve çekirdekli eritrositleri, sıtma ve babesioziste çekirdek içi parazitleri ve ağır demir eksikliği anemisinde kalem şeklindeki deformiteleri içerir. Ayrıca miyeloid hücre ve trombositlerin incelenmesi de tanıya yardımcı olur. Hipersegmente nötrofiller ve büyük trombositler megaloblastik anemi tanısını destekler ve olgunlaşmamış blast formlarının varlığı, lösemi tanısını koydurabilir. Hipoplastik anemilerin tanısında ortalama eritrosit hacmi (MCV), oldukça yararlı bir değerdir. Anemik ve retikülosit sayısı düşük bulunan hastalarda eritrosit büyüklüğü, aneminin mikrositik (MCV <80), normositik (MCV 80- 100) veya makrosirik (MCV >100) olup olmadığını belirlemede kullanılır. Hipoplastik aneminin eritrosit büyükıüğüne dayalı ayırıcı tanısı, bir sonraki bölümde anlatılmaktadır. Retikülosit sayısı yüksek olan anemik hastalarda yeni eritroid hücrelerin aşırı üretimi, kemik iliğinin normal olduğunu ve anemi stresine uygun bir cevap verdiğini düşündürür. Bu durumda kemik iliği incelemesi, nadiren gereklidir çünkü kemik iliğinde genellikle primer anomali patolojisi olmaksızın yalnızca eritroid hiperplazi sergilenecektir. Eritrosit tüketimine neden olan durumun, kanama veya hemoliz olup olmadığı yönünde araştırılma yapılmalıdır. Buna karşılık hipoplastik aneminin değerlendirilmesi için genellikle kemik iliği incelemesi gerekir. Demir eksikliği anemisi gibi sık görülen anormalliklerin dışlandığı hastalarda, kemik iliği infiltrasyonu, kemik iliğinin katıldığı granülamatöz hastalıklar, kemik iliği aplazisi veya myelodisplazi gibi anormallikleri araştırmak için biyopsi endikasyonu vardır.
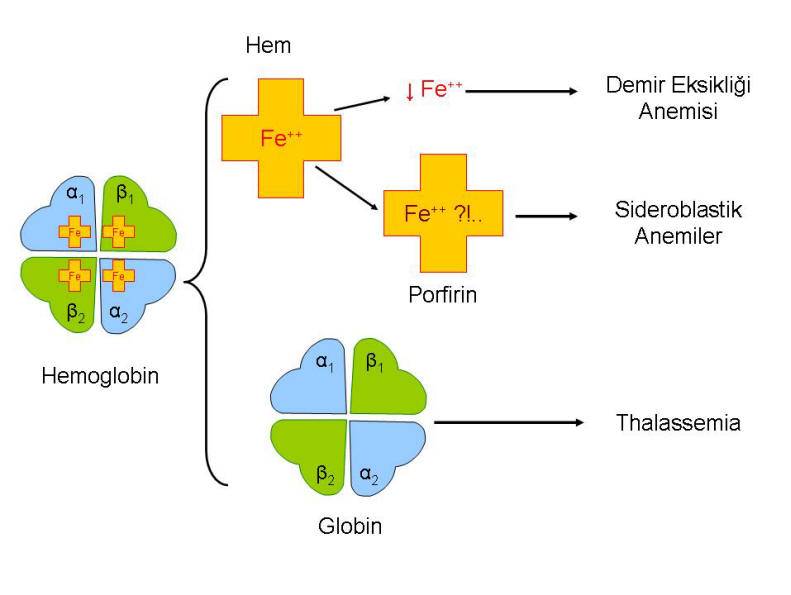
Mikrositik aneminin ayırıcı tanısı özetlenmektedir. Hem sentezinde yetersizlik ya da globin yapımında bozukluklara bağlı oluşabilen hemoglobin sentezindeki defektIerin yol açtığı anemilerin temel özelliği, mikrositoz ve hipohomidir. Mikrositik aneminin başlıca nedeni, porfirin halkasına katılacak demirin olmamasına bağlı olarak hem sentezi yetersizliğine yol açan demir eksikliğidir (demir eksikliği anemisi bir sonraki bölümde ayrıntılı olarak anlatılmaktadır). Kronik hastalığa bağlı anemilerin % 30’una yakınında mikrositoz bulunur. Kurşun zehirlenmesi, demirin heme katılmasını engelleyerek mikrositik anemiye neden olur. Sideroblastik anemi, porfirin halkası sentezindeki bozukluktan kaynaklanır, bu da genellikle hemosentez yolu enzimlerinin inhibisyonu sonucunda oluşmaktadır. Konjenital sideroblastik anemi, hemosentez yolundaki enzimlerden bazılarının kofaktörü olan piridoksine cevap verebilir. Edinsel sideroblastik anemilerin çoğunun nedeni, alkol bağımlılığıdır, çünkü etanol, hem protein sentezindeki enzimlerin çoğunu inhibe eder. Globin sentezindeki yetersizlik, bu bölümün ‘Hemoglobinopatiler’ başlıklı bölümünde ayrıntılı olarak inceleneceği gibi, talasemi sendromlarında görülür. Tüm bu bozukluklar, ortalama korpusküler hemoglobin konsantrasyonunda azalmaya, hipokromi ve eritrosit boyutunda küçülmeye (MCV düşüklüğü) neden olurlar.
Demir eksikliği, bütün dünyadaki anemilerin en önde gelen nedenidir. Klasik demir eksikliği, nlihositer anenli olarak kendini gösterir ancak erken dönemde normositik anemi olarak görülebilir. Bu nedenle anemisi olan tüm hastalarda demir eksikliği düşünülmeli ve yapım azlığına bağlı anemilerde, MCV’den bağımsız olarak demir göstergelerinin ölçülmesi, laboratuar incelemelerinin bir parçası olmalıdır. Demir, hem içeren (ette bulunur) ve hem içermeyen (ıspanak gibi sebzelerde bulunur) besinlerden elde edilir. Hem kaynaklı demir, diğerine göre daha iyi emilir. Demir eksikliğinde ve inefektif eritropoezi olan hastalarda demir emilimi artar. Demir, transferi n reseptörüyle eritrosit prekürsörleri tarafından alımına aracılık eden transferine bağlı olarak ince barsağın proksimal kısmından emilir. Daha sonra demir ayrılarak heme katılır. Hemoglobin üreten hücreler dışındaki demir, fen-itinde depolanır. Erkek ve kadınlarda sırasıyla kg başına toplam 50 ve 40 mg/kg demir bulunur. Toplam demirin % 60 ile 70’i hemoglobinde bulunur. Küçük bir miktan (2mg/kg) hem içeren ve içermeyen enzimlerde, Smg/kg’ı miyoglobinde bulunur. Demirin geri kalanı ise başlıca karaciğer, kemik iliği, dalak ve kasta bulunan ferritinde depolanır. Demirin atılım kapasitesi oldukça kısıtlıdır ve gastrointestinal aşırı emilimi olan hastalarda (inefektif eritropoezde veya konjenital hemohomatozisin sonucu olarak) veya kronik transfüzyonlarda aşırı demir birikimi görülür. Demir yükü, bu dokularda artmış demir birikimine ve endokrin organlarda sekonder birikimlere neden olarak karaciğer disfonksiyonu, diyabet ve diğer endokrin anormaııiklere yol açar. Demir eksikliğinin en sık nedeni, gizli kan kaybıdır. Tüm erkek ve postmenopozal kadınlarda demir eksikliği görüldüğünde, gizli kan saptanıp saptanmadığına bakılmaksızın gastrointestinal kanama yönünden inceleme yapılmalıdır. Premenopozal kadınlarda demir eksikliği, çoğunlukla menstruasyon (yaklaşık 15 mg/ay) ve gebelik sırasındaki (yaklaşık 900 mg) demir kaybına bağlıdır.
Nutrisyonel demir eksikliği en çok, büyümeye oranla demir alımının yetersiz kaldığı çocuklarda ve demir içeren besinler yerine daha çok süt içen bebeklerde görülür. Laboratuar İncelemeleri. Daha önce de belirtildiği gibi, demir eksikliğinin erken döneminde, klasik demir eksikliğinin karakteristiği olan mikrositoz ve hipohomi görülmez. İleri dönemdeki demir eksikliğinde kan yaymasında genellikle hipokromik eritrositler, target ve kalem şeklinde eliptik hücreler gözlenir. Demir eksikliğinin erken döneminde genellikle reaktif trombositoz görülür. Demir eksikliğinin tanısının temelini, periferik kandaki demir indeksleri oluşturur. Bunlar, demir, toplam demir bağlama kapasitesi (TIBC, TDBK) ve territindir. Transferrin saturasyonu, serum demirinin transferrin konsantrasyonuna oranıdır ve normali en az %20’dir. Demir eksikliği, serum demirinde azalmaya, demir bağlama kapasitesindeki artışa ve sonuç olarak transferin saturasyonunun % 10’un altına düşmesine yol açar. Kronik enflamatuvar durumlarda (örneğin enfeksiyon, enflamasyon, malignite), demir ve demir bağlama kapasitesi azalır fakat transferin saturasyonu genellikle % 20’nin üzerinde kalır. Ferritin düzeyi, toplam vücut denıir deposunun bir göstergesidir. Karaciğer, toplam vücut demir miktarına göre ferritin sentezi yapar, 12 ng/ml’nin altındaki bir ferritin düzeyi, demir eksikliği tanısını büyük oranda destekler. Ancak ferritin bir akut faz reaktanıdır ve ateş, enflamatuvar hastalık, enfeksiyon ve diğer stresIerde yükselir. Bununla birlikte strese cevap olarak ferritin düzeyi artışı 50 ile 100 ng/ml’nin üzerine çıkmadığından, 100 ng/ml’nin üzerindeki ferritin düzeyi geneııikle demir eksikliğini dışlar. Demir profilinin indirekt ölçümü, demir eksikliği tanısının doğrulanması veya dışlanmasında yetersiz kalırsa, kemik iliği demir depolarını değerlendirmek amacıyla kemik iliği incelemesi yapılabilir. Kemik iliğinde demir olması, demir eksikliğini dışlar çünkü demir eksikliğine bağlı eritrosit üretimi azalmadan önce kemik iliğindeki demir deposu tükenir. Bunun aksi durum olan kemik iliğinde hiç demir olmaması ise demir eksikliği tanısını destekler. Tedavi. Demir eksikliği, günde iki veya üçe bölünmüş dozlarda ferröz sülfat veya terröz glukonat ile yapılan oral demir takviyesi ile tedavi edilir. Hastalar tedavi sırasında demir preparatlarına bağlı diyare veya kabızlıktan şikayet edebilir, bu durumda semptomatik tedavi uygulanır.
Dozun azaltılması ve kademeli olarak tam doza geçilmesiyle oral tedavi sürdürülebilir. Aneıninin düzeltilmesinden birkaç ay sonrasına kadar, demir depolannın doldurmak amacıyla tedaviye devam edilmelidir. Emilim bozukluğu olan hastalarda, oral demire toleransın olmaması veya oral suplementasyonun demir ihtiyacını karşılamada yetersiz olması durumunda, parenteral demir verilebilir. Özellikle demir dekstran tedavisinde olmak üzere, parenteral demir uygulamasında anafılaksi görülebilir. Ancak sodyum ferrik glukonat ve demir sühoz gibi yeni çıkan preparatlar önemli oranda daha güvenlidir. Daha önce de belirtildiği gibi, tüm erkek ve menopoz sonrası dönemdeki kadınlarda görülen demir eksikliğinde, gastrointestinal kanama nedenleri araştırılmalıdır.
Yapım azlığına bağlı mahositik aneminin iki kategorisi vardır: megaloblastik anemi ve megaloblastik olmayan makrositik anemi. Megaloblastik anemi, DNA sentezinin yetersizliğinden kaynaklanır ve buna bağlı olarak tüm hızlı bölünen hücrelerin sitoplazma ve çekirdeğinin eş zamanlı olmayan olgunlaşmasıyla sonuçlanır. Megaloblastik olmayan makrositik anemiler genellikle, kolesterol metabolizmasındaki anormalliklerden kaynaklanan membran bozukluklarının göstergesidir ve en çok, ileri dönem karaciğer hastalığı veya ağır hipotiroidisi olan hastalarda görülür. %10’un üzerindeki retikülositoz oranı, otomatik kan sayımında MCV yüksekliğine neden olur çünkü retikülositler, olgun eritrositlerden daha büyüktür.
Megaloblastik anemi, hücre döngüsünün S fazında durmasına yol açan DNA 'nın önemli nükleotid öncülerinin sentezinin engellenmesinden kaynaklanır. Bu anemide sitoplazmik olgunlaşma meydana gelir ancak çekirdeğin olgunlaşması durur. Büyük ve olgunlaşmamış bir çekirdeği çevreleyen olgun görünümlü sitoplazmasıyla bu hücreler tuhaf bir görüntüye sahiptirler. DNA sentezinin engellenmesi, hızla bölünen bütün hücreleri etkilediği nden megaloblastik anemisi olan hastalarda çoğunlukla pansitopeni ve diyare ve/veya malabsorbsiyon gibi gastrointestinal semptomlar görülür. Kadınlarda, servikal mukozada megaloblastik değişiklikler olur ve anormal Papanicolaou smear alarmı verebilirler. Megaloblastik aneminin en sık nedenleri arasında BI2 vitamini veya folat eksikliği, DNA sentezi veya folat metabolizmasını engelleyen ilaçlar ve mjyelodisplazi bulunur. BI2 Vitamin (Kobalamin) Eksikliği. Kobalarrun (B 12), besinlerdeki hayvansal proteinden emilir. B 12metabolizma ve emilim süreci karışıktır çünkü B 12daima diğer proteinlere bağlıdır. Proteine bağlı vitaminler midede pepsinle sindirilerek serbest hale geçerek transkobalamin I’ya bağlanır. R bağlayıcılar olarak adlandırılan transkobalamin I ve III, hızlı elektroforetik mobilitelerinden dolayı tüm salgılarda, plazmada ve nötrofillerin sekonder granülleri arasında saptanırlar. R bağlayıcı/arın B12 depolanmasında rol oynadığı varsayılsa da, işlevleri bilinmemektedir ve izole konjenital eksiklikleri klinik olarak sessizdir. Proksimal duodenumda pankreatik proteazlar B12’yi sindirerek R bağlayıcılardan ayırır, B12bundan sonra intrensek faktöre (IF) bağlanır.
IF, midenin parietal hücrelerinden salgılanır ve distal ileumda IF spesifik reseptörleri ile B12’runemilmesine aracılık eder. İleumdaki mukoza hücrelerinde tekrar sindirilen IF-B 12 kompleksinden B 12aynlarak plazmaya bağlı transkobalamin II ‘e (TK II) bağlanır, bu taşıyıcı protein B 12’ninTkll spesifik reseptörleri ile hücre içine girişine aracılık eder. B12,hücre içinde metilmalorul-koenzim A (CoA) mutaz ve homosistein-metionin metiltransferaz adlı iki hücre içi enzimin kofaktörüdür. Metilmalonil-CoA mutaz, sitrik asit siklusunda metilmalonil-CoA’yl suksinil CoA’ya dönüştürme işlevini gören mitokondriyal bir enzimdir. Sitoplazmik enzim olan homosistein-metionin metiltransferaz, metionin oluşturmak üzere metil gruplarının, N-metiltetrahidrofolattan homosisteine transfer edilmesi için gereklidir. Demetile tetrahidrofolat, deoksiüridinin deoksitimidine dönüşümü için bir karbon vericisi olarak gereklidir. B 12’ninolmaması, tetrahidrofolatın metile formunda kalmasına yol açar ve DNA’yı oluşturacak timidin S’-trifosfat sentezi engellerur. Bu fonksiyonel folat eksikliği, B12 eksikliği tarafından indüklenen megaloblastik değişikliklerin nedenidir ve B 12ve folat eksikliğinin indüklediği hematolojik anormalliklerdeki benzerliği açıklar.
Kobalamin Eksikliğinin Nedenlerİ. BI2 eksikliğinin en sık nedeni, mide parietal hücre atrofisi, gastrik asit salgısı bozukluğu ve IF eksikliğiyle ilişkili otoimmun bir hastalık olan pernisiyöz anemidir. Antiparietal hücre ve anti IF antikorları, pernisiyöz anemili hastalarda sık bulunur ve Graves hastalığı, Addison hastalığı ve hipoparatiroidizm gibi diğer otoimmün hastalıklarla da ilişkilidir;.Gastrointestinal sistemdeki diğer pek çok lezyon, B 12’nin emilimini bozabilir. Gastrektomi parietal hücre fonksiyonu ve IF salgısının kaybına yol açar. Pankreas yetersizliği, R bağlayıcl-Bl2 kompleksinin sindirimini bozarak Bl2’nin IF’ye bağlanıp ileumdan emilmesini engeller. Terminal ileumun rezeksiyonu,Crohn hastalığı, sprue, barsak tüberkülozu ve lenfoma gibi ileumun mukoza fonksiyonlarını etkileyen diğer hastalıklarda olduğu gibi, B 12vitamininin emilmesini engeller. Bl2’nin deposu büyük ve günlük kaybı da düşük miktarda olduğundan, alııru aniden kesilse bile depolar 3-4 yıl yeterli olur. Bu nedenle B 12emilim bozukluğu birkaç yıl sürmedikçe, eksiklik belirtileri ortaya çıkmaz. Nutrisyonel B 12eksikliği nadirdir ve yalnızca, yıllarca tüm hayvansal ürünlerin kısıtlandığı katı vejetaryan diyetle beslenenlerde görülür. Vejetaryan annelerin emzirdiği bebekler de B12eksikliği bakıırundan risk altındadır. Folat, yeşil yapraklı sebze meyve gibi bitkisel yiyeceklerde ve hayvansal proteinlerde olmak üzere, besinlerde bolca bulunur. Ancak uzun süreli pişirme sırasında etkisi azaldığından, taze sebze ve meyveler, en güvenilir folat kaynaklarıdır. Bu nedenle taze sebze ve meyveden fakir beslenen kişilerde nutrisyonel folat eksikliği çok yaygındır. Folat eksikliği, gebelik, hemoliz ve eksfolyatif dermatit gibi gereksinirnin arttığı ve diyaliz gibi kaybın arttığı durumlarda da meydana gelebilir. Folat emilimi ince barsağın proksimal bölümünde gerçekleşir ve emilimdeki bozukluk folat eksikliğine yol açabilir.
İlaç ve toksinler, megaloblastik aneminin en sık görülen nedenleridir. Metotreksat ve sulfonamidler gibi bazı ilaçlar, doğrudan folat antagonisti gibi etki ederek folat eksikliğini taklit ederler. Pürin ve pirimidin analogu kemoterapötik ajanlar (örn., azatiyopürin, 5-florourasil) doğrudan DNA sentezi inhibitörleridir. Antiviral ajanlar, açıklanamayan bir mekanizmayla megaloblastik değişikliklere yol açarlar. Alkol, folat metabolizmasını bozar ve sıklıkla eşlik eden folat eksikliğinin etkisi artırır. Myelodisplazi, başlıca eritroit serideki megaloblastik değişikliklere neden olarak makrositik anemi şeklinde kendini gösterir.
MegalobIastik aneminin gelişimi genellikle çok yavaş olduğundan hipovolemiyi önleyecek plazma artışı için yeterli zaman kalır. Bu nedenle hastalarda klinik belirti verdiğinde çoğunlukla aneminin derecesi şiddetlidir. Hastalar, solukluk ve sarılığın birleşimi sonucunda sarımsı bir cilde sahiptir. Bazılarında glosit ve şeliozis vardır. Ciddi anemili hastalarda MCV 100’ün üzerindedir, ancak barsaklardaki megaloblastik değişikliklerin neden olduğu ek demir eksikliğinin olması, makrositozda azalmaya neden olabilir. Hastaların çoğunluğunda pansitopeni vardır. Periferik yaymada büyük, oval hücreler (makroovalositler), hipersegmente nötrofiller ve büyük plateletler görülür. Kemik iliği, megaloblastik değişiklikler ve anormal derecede büyük öncül hücreler ile hiperselüler özellik gösterir. Ayrıca ciddi inefektif hematopoez ise eritrositlerin intramedüller yıkııru neticesinde oluşan yüksek bilirübin ve laktat dehidrojenaz ile kendini gösterir. B12eksikliğinde, megaloblastik aneminin diğer nedenlerinde görülmeyen nörolojik anormallikler olabilir. Nörolojik belirtiler, arka kolon demyelinizasyonunun neden olduğu subklinik vibrasyon ve pozisyon duyu su kaybından aşikar demans ve nöropsikiyatrik hastalığa kadar geniş bir yelpazede olabilir. B12eksikliği olan hastalar, özellikle megaloblastik anemilerin hematolojik bulgularıru düzelten, ancak nörolojik anormaHiklerini tedavi etmeyen folat ile tedavi edildiklerinde, anemi olmaksızın sadece nörolojik değişiklikler gösterebilirler. B12eksikliğinin nörolojik belirtilerinin, metilmalonil CoA mutaz mitokondriyal enzim işlevinin kaybından kaynaklandığı düşünülmektedir. Metabolizması bozuk tek zincirli yağ asitlerinin miyeline katılmasının nörolojik disfonksiyona neden olduğu öne sürülen açıklamalar arasındadır ve bu bulguların neden yalnızca B 12eksikliği olan hastalarda görülüp, neden folat yolundaki anormalliklerden kaynaklanan megaloblastik anemisi olanlarda görülmediğini açıklamaktadır.
Periferik kanda BI2 ve folat ölçümü megaloblastik anemi tanısını doğrular. Barsak mukozasındaki megaloblastik değişiklikler, B 12 eksikliği olduğunda, folat emilim bozukluğunun eşlik ettiği veya tam tersi duruma yol açabildiği için megaloblastik hematopoezde her iki ölçümün de yapılması gereklidir. Serum folatının normal olduğu fakat klinik olarak eksikliğinden şüphelenildiği durumlarda eritrosit folat düzeyleri vücut folat depolarının daha iyi bir göstergesidir. B 12ve folat eksikliğinde homosistein düzeyleri yükselir, B 12eksikliğinde metilmalonik asit seviyesi artar. B12eksikliğinden şüphelenildiği ve serum seviyelerinin normal aralıkta olduğu durumlarda bu testler istenebilir. B12 eksikliğinin nedenini belirlemede Schilling testinden yararlanılabilir. Bu test için, oralolarak radyoaktif B12, çok miktardaki işaretlenmemiş B12ile birlikte, parenteral yoldan verilir. Daha sonra, idrarla atılan radyoaktivitenin ölçülmesiyle oral verilen Bl2’nin emilimi saptanır. IF’ye bağlı ve farkl i bir izotopla işaretlenen B 12eşzamanlı olarak verilebilir. IF’ye bağlı B l2’nin selektif olarak emilmesi, pernisiyöz anemi tanısını destekler. İzotopların hiçbirinin emilmediği durumda, potansiyel bakteri üremesini tehdit eden antibiyotikler veya pankreatik yetersizliğini dışlamak amacıyla pankreas enzimlerin verilmesini takiben test tekrarlanabilir. Günümüzde antiparietal hücre antikor ve anti-IF antikor düzeylerinin ölçülebilmesi ve özellikle yeterli idrar toplama gerektirmesi testin güvenirliliğini etkilemesi nedeniyle, Schilling testi giderek daha az kullanılmaktadır.
Megaloblastik anemisi olan hastaların hematokriti sıklıkla çok düşük düzeydedir. Buna bağlı olarak, hastalık düşünülüyorsa, folat ve B 12 düzeyleri için kan örneği alındığında hemen tedaviye başlanmalıdır. B 12 eksikliğinde, 7 gün boyunca intravenöz yoldan günde 100 mcg parenteral tedavi başlatılmalıdır. Ayda bir defa 1 mg’lık intramusküler yoldan yapılan tedavi başka bir alternatiftir. Kronik tedavi ise ayda bir defa 1 mg şeklinde uygulanır. Nutrisyonel B 12 eksikliği olan hastalar oral replasman tedavisi alabi iir1er. Yüksek doz kristal ize B i2 vitamini ile yapılan oral tedavi, normal B 12 emilimin blokajını engelleyebilir bu nedenle parenteral tedaviyi reddeden hastalarda oral tedavi bir seçenek olabilir. Bu tedaviye ek olarak, beraberinde sekonder folat eksikliği de bulunabileceğinden dolayı tedaviye folat da eklenmelidir. Folat eksikliği olan hastalara günde 1-5 mg folat replasmanı yapılabilir. Daha önce de belirtildiği gibi, hastaların B12 eksikliğinin dışlanması gereklidir çünkü folat replasmanı, B12 eksikliği olan kişilerde hematolojik parametreleri düzeltir ancak nörolojik sekeli ortadan kaldırmaz. Megaloblastik tedavinin başlamasını takiben çok hızIı bir cevap alınır. Tedaviyi takiben 2 gün içinde retikülositoz görülür ve 7- 10 gün içinde pik yapar. Nötropeninin hızla düzelmesine karşın, nötrofil hipersegmentasyonu günlerce sürebilir. Bu süreç içinde hızlı hücre yapım ve döngüsü meydana gelerek hipokalemi, hiperürisemi veya hipofosfatemiye zemin hazırlayabilir. Replasman cevabında hızlı hücre yapımı demir ihtiyacını artırabileceğinden hastalar demir eksikliği gelişimi açısından takip edilmelidir. Anemi ve diğer sitopeniler, tedaviye bir iki ay içinde tamamen cevap verirler ancak B 12 eksikliğinin nörolojik bulguları oldukça yavaş düzelebilir veya tamamen geri dönüşümsüz olabilir.
Normositik hipoplastik aneminin ayırıcı tanısı, oldukça ayrıntılıdır. Mikrositoz veya makrositoza neden olan nutrisyonel anemilerin çoğu, normositik anemi şeklinde başlar. Kombine nutrisyonel anemilerde de MCV normal olabilir. Normositik anemilerin tanısında EPO düzeylerinin ölçülmesi yararlı olabilir. Böbrek yetersizliğinden kaynaklanan anemilerin tanısında da yardımcı olur. Kronik enflamasyon ve endokrinopatilere bağlı anemilerin pek çoğunda EPO düzeyleri düşer. Ancak hematokrit düzeyi %30’un altına düşmedikçe EPO düzeyi normal aralığın üstüne genelikle çıkmadığından, hafif anemik olgularda EPO değerlerinin yorumlanması zor olabilir. Hematokrit düzeyinin %30’un altına düştüğü durumlarda bile genellikle EPO değerleri normalolabilir fakat anemi tablosunda bu düzeyler uygunsuz olarak düşüktür. Yüksek EPO düzeyleri, kemik iliğinin anemiye cevabının yetersizliğini düşündürür ve miyelofitiz veya primer kemik iliği yetersizliği olasılığını artırır. Rutin endokrin ve nutrisyonel incelemelerle tanının açıklanamadığı durumlarda, primer kemik iliği patolojilerini dışlamak amacıyla, kemik iliği incelemesi yapılmalıdır.
Kronik hastalık anemisi, kronik enflamatuvar, enfeksiyöz, malign ve otoimmün hastalığı olanlarda görülür. Kesin veya relatif EPO yetersizliği, eritrapoezin direkt inhibisyonu, gelişmekte olan eritrasitlere yetersiz demir girişi ve/veya eritrosit ömrünün kısalması, kronik hastalık anemisine neden olur. Hastaların serum demir düzeyleri düşüktür fakat demir eksiliğindeki profilin tersine, burada, demir bağlama kapasitesi de azalmıştır ve transferin saturasyonu % 10’un üzerindedir. Akut faz reaktanı olması ve azalmış demir katılımı nedeniyle, ferritin düzeyi, genellikle yüksektir. Kronik renal yetersizliğe bağlı aneminin temel tedavisi, EPO replasmanıdır. Kronik hastalık anemisi, altta yatan kronik hastalık tedavi edilirse düzelir. Ancak primer tedavi yapılmadığı durumda, bu anemilerin EPO tedavisine genellikle cevap vermesi nedeniyle, hangi hastaların tedaviye cevap verme olasılığı olduğunu belirlemede EPO tedavisi yararlı olur. EPO tedavisi 150 ünitenin altındaki düzeylerde başarılı olabilir, bununla birlikte en fazla başarı, 50 ünitenin altındaki düzeylerde sağlanır. EPO replasmanına dramatik yanıt en çok, özellikle multiple miyelom gibi
bazı malignite, romatoid artrit ve HIV enfeksiyonuna bağlı anemisi olan hastalarda alınır. Normositik aneminin diğer nedenlerinin tedavisi, hastalığın primer kaynağına bağlıdır. Primer kemik iliği yetersizliği sendromları ve hematolojik malignitelerin teşhis ve tedavisi anlatılmıştır.
Anemi tablosunda retikülosit sayısının artması, olgunlaşmamış eritrosit kaybına normal bir kemik iliğinin kompansatuvar cevabını gösterir. Hemoliz, eritrositlerin retiküloendotelyal sistem (ekstrensek hemoliz) veya kan damarlarında (intrensek hemoliz) olgunlaşmadan tahrip olması demektir. Retikülositozun görüldüğü anemilere neden olan tek diğer durum, akut kanamadır. Hemolitik aneminin ayırıcı tanısı özetlenmiştir. Periferik yayma, aneminin tanımlanmasında sıklıkla yardımcı bir yöntemdir ancak hemolitik anemisi olan hastalarda özellikle kritik önem taşır. Daha önce de belirtildiği gibi, eritrositlerin morfolojik incelemesi, immün hemolizin mikroanjiopatik hemolizden ayırt edilmesinde yararlıdır. Ayrıca, eritrosit morfolojisindeki diğer anormallikler, orak hücre hastalığı (oraklaşmış hücre), enzim defektIeri (bire cel!) veya eritrosit membran bozuklukları (sferosit ve eliptositler) gibi spesifik hastalıklar için karakteristiktir.
İmmün kökenli hemoliz, eritrosit membranı dış yüzeyinin antikor ve veya komplementle kaplanmasından kaynaklanır. İmmünglobin G (IgG) (sıcak antikarlar) veya immünglobin M (lgM) antikorları (soğuk antikarlar) ile oluşabilir. Sıcak ve soğuk terimleri, antikorların maksimum bağlandığı dereceye göre belirlenmektedir. Bu her iki antikor tipinin neden olduğu klinik sendromlar belirlidir. Hemolitik aneminin tanısı, direkt ve indirekt antiglobulin (Coombs) testine dayanır. Direkt Coombs testinde, hastanın eritrositleri tavşan antiserumuyla karıştırılarak insan IgG veya insan komplementi ile karşılaştırılır. Daha sonra izlemeye alınan hücrelerde aglutinasyon olması, hastanın eritrositlerinde antikor ve/veya komplementin varlığını gösterir. İndirekt Coombs testi, hastanın serumunun ABO-uyumlu eritrositlerle karıştırılması ve bu karışımın IgG’ye karşı oluşturulmuş tavşan antiserumuyla işleme tabi tutulmasıdır. Coombs testi, hastanın serumundaki antikor varlığının saptanmasını sağlar.
Klasik otoimmün hemolitik anemiye (KOHA), eritrosit antijenlerine karşı oluşan IgG antikoru neden olur. Sıcak antikorlar ile oluşan hemoliz, primer (idiyopatik) olabilir veya otoimmün hastalık, lenfoproliferatif hastalıklar veya ilaçlara bağlı gelişebilir. Hastalarda anemi, sarılık ve artmış retikülosit sayısı görülür. Bazı hastalarda splenomegali vardır. Pozitif Coombs testinde gösterildiği gibi, eritrosit membranında IgG’nin varlığının ortaya konmasıyla, tanı, laboratuar olarak doğrulanır; bazı hastalarda kompleman da bulunur. Antikorların retikülosit ve olgun eritrositleri parçaladığı nadir hastalarda, retikülositoz görülmez. KOHA’nın temel tedavisi, kortikosteroidlerdir. Hastalara genellikle 1-2 mg/kg prednizon tedavisi başlanır ve cevap alınmasını takiben birkaç ay içinde yavaş bir şekilde azaltılarak kesilir. Prednizona cevap vermeyen hastalarda,siklofosfamid, azatiyoprin veya klorambüsil gibi diğer immünsupresif ajan tedavileri uygulanabilir. Bazı hastalar LVimmünglobuline cevap verebilir. Steroide dirençli veya cevapsız bazı hastalarda splenektomi etkili olabilir ancak splenektomi sonrası cevapsız ve hemolizi devam eden hastaların sekonder tromboembolik olaylar açısından yüksek risk taşıdığı yönünde kanıtlar vardır. Sıcak antikorlar, daca bağlı hemolize neden olurlar. İlaçların KOHA’ya neden olması için birkaç mekanizma
vardır. Penisilin eritrositlere bağlanarak hemoliz oluşturur ve hapten gibi hareket eder; ilaca karşı antikor oluşur ve yalnızca ilaç varlığında hemoliz meydana gelir. Tip 2 hemolize, eritrosit membranına bağlanan ve komplemanı aktive eden bir antikor-ilaç kompleksi oluşmasıyla meydana gelir. Bu tip hemolize neden olan ilaçlar, kinidin, kinin ve rifampindir. Metildopa ve prokainamidin dahil olduğu bazı ilaçlar, Rh ve diğer eritrosit antijenlerine karşı gerçek antieritrosit antikor oluşmasını indükleyerek hemolize neden olurlar. Antikorlar, ilaç olmadığında da varlığını sürdürebilir fakat pozitif bir Coombs testi bulunan hastaların hepsinde hemoliz görülmeyebilir.
Soğuk antikorlar ile oluşan immün hemoliz, genellikle postenfeksiyözdür. En sık eşlik eden hastalık nedenleri, Mycoplasma pneumoniae ve Epstein Barr virusudur (EBV). IgM antikorları, eritrosit antijen i (Mycoplasma) veya (EBV) antijenine karşı yapılır. Daha düşük sıcaklıklarda ve genellikle periferik dolaşırnda antikorlar eritrosit ve komplemana bağlanır. IgM, santral dolaşıma ulaştığında eritrositten ayrılır ve komplemana bağlı kalır. Bu nedenle Coombs testi, IgG veya IgM için negatif, kompleman için pozitif olur. Hemoliz kendini sınırlayan özelliktedir ve nadiren şiddetli olur ve destekleyici tedavi ile düzelir. Transfüzyon gerektiren ağır hemoliz olgularında, daha fazla hemolizi engellemek için kan, ısıtıcı ile verilmelidir. Soğuk aglutinin hastalığı, genellikle lenfoproliferatif hastalıklarla birlikte görülen kronik bir IgM aracılı hemolizdiL Ayrıca, nadiren şiddetli olsa bile soğuk aglutinin hastalığı, genellikle düşük dereceli kronik hemolize neden olur. Bu hastalar, steroid ve splenektomiye az cevap verirler. Akut ciddi TgMaracılı hemoliz, plazmafereze cevap verebilir. Soğuğa maruz kalmayı önleme, destekleyici tedavidir. Lenfoproliferatif hastalıkta, rituximab (anti- CD20) ile yapılan immün tedaviye cevap alınabilir.
Mikroanjiopatik hemolitik anemi (MAHA), eritrositlerin küçük damarlardan geçtiği sırada travmatik olarak harabiyetine bağlıdır. MAHA’nın önde gelen nedenleri arasında, dissemine intravasküler koagülasyon (DIC) ve trombotik trombositopenik purpura/hemolitik üremik sendrom (TTP/HUS) vardır. Diğer nedenler arasında, preeklampsi, eklampsi ve HELLP sendromu (preklampsiye eşlik eden hemoliz, karaciğer enzim yüksekliği trombosit düşüklüğü) gibi gebelikle ilişkili sendromlar, ilaçlar ve metastatik kanserler bulunur. Benzer bir hemolitik tablo da, hasarlı bir kalp kapağında meydana gelen travmatik hemolizde görülebilir. Periferik yaymada siztositlerin (parçalanmış eritrosit) varlığı, MAHA tanısını destekler. Protrombin zamanı ve parsiyel tromboplastin zamanının normal olması, DIC yerine TTP/HUS tanısını destekler.
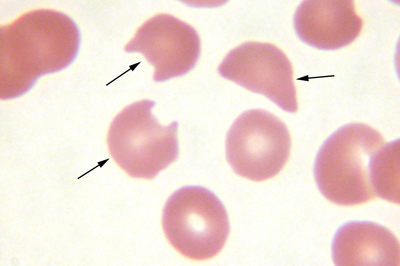
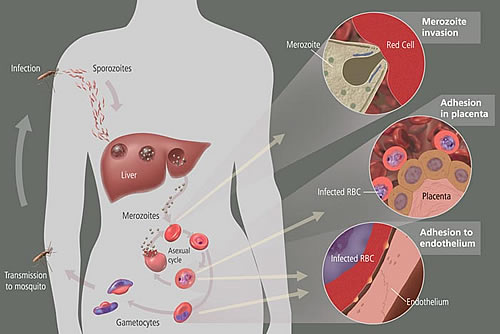
Eritrositlerin, malarya, babesiozis ve bartonellozisde olduğu gibi parazitlerle doğrudan enfeksiyonuyla hemoliz meydana gelebilir. Bakteriyel toksinierin membranı doğrudan etkilediği klostridiuma bağlı sepsisde, ağır hemoliz görülebilir. Kalıtsal Sferositoz (HS), eritrosit hücre iskeletindeki proteinlerde görülen çeşitli konjenital anomalilerden kaynaklanır. HS’lu hastaların çoğunda, spektrin veya ankyrinde dominant kalıtsal mutasyonlar bulunur. HS, hemolitik anemi, splenomegali ve periferikkanda belirgin sferositlerin varlığıyla tanımlanır. Sferositler, bozuk hücre iskeleti sonucunda oluşan anormal membran parçalarının retiküloendotelyal sistem hücreleri tarafından dalakta tutulması sonucunda eritrositlerin yeniden şekillenmesidir. Sferositler, membran sitoplazma oranını düşüren dalakta membran kaybını gösterir. Membran/sitoplazma oranının yüksekliği, normal eritrositin esnek ve bikonkav şeklini anlatır. Membranını kaybeden eritrosit bikonkav morfolojik özelliğini yitirerek sferotik bir şekil alır. Sferositler daha az esnektir ve kılcal damarlarda tahrip olabilir. HS’nin karakteristik laboratuar bulgusu, eritrositlerin yüzey membranının azalmasının genişleme özelliğini kaybettirmesi ve buna bağlı ozmotik frajilitenin artmasıdır. HS, genellikle iyi kompanse edilen hafif bir hastalıktır. Hastalarda tipik olarak, enfeksiyonlar sırasında veya kemik iliğini baskılayan ilaçlar aldıklarında alevlenmeler görülür. Belirgin hemolizi olanlara folat desteği verilmelidir. çoğu hastada pigment taşları için kolesistektomi gerekir. Ağır, semptomatik anemi, splenektomi ile tedavi edilir. Herediter eliptositoz (HE), genellikle membran proteinleri ve altındaki sitoplazmik proteinleri etkileyen dominant kalıtsal mutasyonlardan oluşur. En sık olarak, eritrositlerin eliptik şeklinden sorumlu spektrin ve protein 4.1 'in etkileşimini bozan anormallikler görülür. HS’da olduğu gibi bu hastaların hafif hemoliz ve splenomegalisi vardır. Herediıer propoikilositozis (HPP), genellikle, 2 ayrı membran bozukluğunun kalıtımınına (örn., HS için bir alel, HE için bir alel) bağlı nadir görülen resesif bir hastalıktır. Hastaların kan yaymasında mikrosferositoz ve eliptositler bulunan çok daha ağır hemoliz vardır. HS’da olduğu gibi HE ve HPP’deki semptomatik aneminin tedavisi splenektomidir
Paroksismal Noktürnal Hemoglobinüri. Paroksismal no ktürnal hemoglobinüri (PNH), kompleman regülasyonundaki bozukluğa bağlı edinsel bir klonal hastalıktır. Normal eritrositler, geç hızlandırıcı faktör (DAF, delay-accelerating factor) ve reaktif lizis membran inhibitörünün (MIRL) de bulunduğu membran proteinleriyle komplemana bağlı hücre lizisinden korunmaktadır. Bu her iki protein de, bir glikozilfosfatidilinositol (GPI) bağıyla membrana bağlı bir protein grubuna aittir. PNH’li hastalarda GPI sentezi için gerekli bir enzim olan fosfatidilinositoglikan A (PIG A)'da klonal mutasyonlar vardır. Bu mutasyonlar hematopoetik kök hücrelerden kaynaklanır ve tüm hematopoetik hücrelerde GPI’ya bağlı proteinler eksiktir. Eritrositlerdeki GPI’ya bağlı proteinlerin olmaması, bu eritrositlerin kompleman la aktive olan lizise karşı hassasiyetini artınr. PNH için klasik testler, asidik serum (Ham testi) veya hipotonik bir ortamda (sukroz lizis testi), eritrositlerin lizise hassasiyet artışına dayalı fonksiyonel testlerdir. PNH tanısı, altta yatan moleküler patolojnin tanımlanmasından sonra yapılan flow sitometre ile eritrosit veya lökositlerin yüzeyinde DAF veya MIRL’nin olmadığının gösterilmesiyle konur. PNH, serbest hemoglobinin salındığı ve hastalığa adını veren hemoglobinürinin görüldüğü epizodik akut intravasküler hemoliz ile tanımJanır. Bu hastalık, myeloproliferatif hastalık spektrumunun bir parçası olarak kabul edilir ve trombotik risk ve lösemi ve/veya miyelofib-röz gelişme riskiyle ilişkili bir klonal kök hücre hastalığıdır.
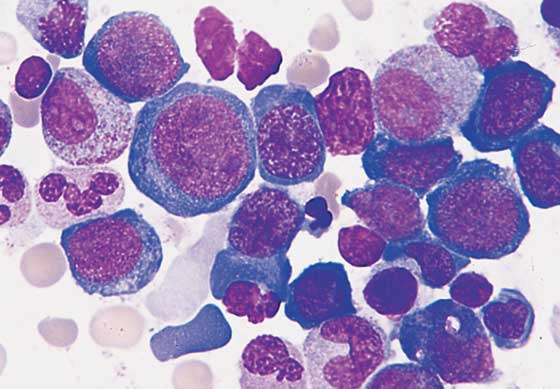
PNH hastaları, Budd-Chiari sendromu, portal ven trombozu ve serebrovasküler tronıboz gibi miyeloproliferatif hastalıklarda görülendekilere benzer tipik trombotik komplikasyonlara hassastır. PNH da aplastk anemiyle ilişkilidir, hastalarda apıazi gelişebilir ve immünsüpresif tedaviye cevap veren apıastik anemili hastalarda sıklıkla PNH benzeri klonlar gelişir. Tedavi, büyük oranda destekleyicidir. Ancak genç hastalarda allojenik kök hücre transplantasyonu düşünülmelidir. Spur Hücreli Anemİ. Spur hücreler (akantositler), ağır karaciğer hastalığı, ağır malnütrisyon, malabsorbsiyon olan ve asplenik hastalarda bulunan anormal morfolojik hücrelerdir. Membranda, anormal lipidler nedeniyle çıkıntılar oluşur. Bu değişiklikler hafif hemolizle ilişkili olabilir ancak ağır karaciğer hastalığı olan hastalarda, hemolizin hipersplenizmden ayırt edilmesi zor olur. Abetalipoproteinemisi olan hastalarda da benzer değişiklikler
gözlenebilir.
Glukoz -6 Fosfat Dehidrogenaz (G6PD), eritrositleri membran ve hemoglobin oksidasyonundan koruyan indirgenmiş glutatyonun hücre içi depolanması için gerekli heksoz monofosfat şant yolunun önemli bir enzimidir. G6PD geni X kromozomlarında bulunur ve hastalığın büyük çoğunlukla erkeklerde görülür. Ancak, çarpık iyonlaşma nedeniyle, nadiren heterozigot bir kadın hastada da eksiklik bulunabilir. çoğu G6PD mutasyonu Afrika ve Akdeniz ülkelerinde görülmekte ve sıtmaya karşı dirençli oldukları için seçildikleri düşünülmektedir. G6PD’nin Afrika formu görece hafif, Akdeniz fonnu ise ağır seyirlidir. G6PD’nin olmaması, eritrositlerin oksidatif strese duyarlı hale getirir. Enfeksiyon, asidoz varlığı veya oksidatif ilaçlara maruz kalındığında hemoglobin hücre içinde presipite olarak hemolize neden olur. Bu hastalıkta, sulfonamid, antimalaryal, dapson, aspirin ve fenasetin dahil pek çok ilaç hemolize neden olabilir. Akut enfeksiyonu olan veya yakın zamanda oksidan ilaçlara maruz kalan hemolizli Afrikan Amerikan veya Akdeniz kökenli erkeklerde bu hastalık da akla gelmelidir. G6PD’nin daha ağır olan Akdeniz formunda, baklaya karşı hemoliz (favizm) gelişebilir. içinde hemoglobin presipite olmuş hücrelerde, kristal viole ile boyanmış periferik yaymasında Heinz cisimcikleri görülür. Bu inklüzyon cisimcikleri, kan yaymasında bite cell görüntüsü vererek dal ak tarafından atılırlar. Periferik kanda G6PD düzeyinin ölçülmesiyle tanı konabilmektedir. Ancak, G6PD’li hastalarda retikülosit ve genç eritrositlerin enzim düzeyi daha yüksektir ve eğer tanı olasılığında, normal düzeydeki G6PD testinin akut epizod geçtikten sonra tekrar edilmesi planlanmaııdır. Bu hastalarda hemolizi önlemenin temel kuralı, oksidatif stresten, özellikle de hemolize yol açma potansiyeli bulunan ilaçlardan kaçınmaktır. Ağır epizodik veya kronik hemolizli hastalarda splenektomi önerilir.
Hemolitik aneminin nadir nedenleri olarak, glikolitik yolda yer alan enzimlerin hemen hepsini içeren eksiklikler bildirilmiştir. Bunlardan en sık görüleni, piruvat kinaz eksikliğidir. Otozomal genler bu enzimleri kodladığından, kalıtım biçimi otozomal resesiftir. Hemoglobinopatiler, anormal enzim senteziyle sonuçlanan mutasyonlardır. Bunlardan en sık görüleni, Orak hücre sendromları ve talasemilerdir ve G6PD gibi, malaryanın endemik olduğu bölgelerde görülür. Orak hücre hastalığı, orak hücre sendromlannın en sık görülenidir. A globin geninin 6. amino asidinde, glutamik asidin valinle yer değiştirmesinden kaynaklanan bir nokta mutasyonudur. Afrika, Hindistan, Akdeniz ülkeleri ve Ortadoğu’daki farklı populasyonlarda birbirinden bağımsız mutasyonlar şeklinde görülmektedir. Hidrofobik yerine hidrofilik parçanın gelmesiyle oluşan mutasyonda, deoksijenize sickle hemoglobin (HbS), suda daha az eriyen özelliğe ve polimerizasyon ve presipitasyona karşı daha duyarlı özelliklere sahip olur. HbS’in çökme hızı, eritrosit içindeki deoksijenize hemoglobin konsantrasyonuna oldukça duyarlıdır. Bu nedenle, hücre hidrasyonundaki değişiklikler (dehidrasyon) veya oksijen disosiyasyon eğrisindeki değişiklikler (örn. hipoksi, asidoz, yükseklik) ve konsantrasyonun arttığı durumlarda oraklaşma artar. Orak hücre hastalığının akut komplikasyonlarından çoğu, vazookluzyona bağlıdır. Organlardaki kılcal damar ağında oklüzyonların meydana getirdiği ve en çok ekstremiteler, göğüs, kann ve sırtta görülen iskemik ağrılar nedeniyle ağrılı krizler görülür. Bu ağrılı krizler, enfeksiyon, dehidratasyon, ani ısı değişiklikleri ve gebelikten etkilenir. Ancak, akut bir ağrılı krizin nedeni genellikle bilinmemektedir. Akciğer dolaşımındaki vazo oklüzyonu, orak hücre hastalığının aşikar bir komlplikasyonu olabilir ve akut göğüs sendromuna yol açabilir. Bu sendrom, göğüs ağrısı, hipoksemi ve göğüs infiltrasyonlarıyla kendini gösterebilir. Akut göğüs sendromunda, enfeksiyon, enfarkt ve insitu trombozların rolü ayırt edilemez ancak bütün hastalar olası bir pnömoni için antibiyotik kullanmalıdır. Çünkü hipoksemi oraklaşma ve solunum sıkıntısını artırdığı için, akut göğüs sendromu, yaşamı tehdit eder ve acil kan transfüzyonu gerektirebilir.
Orak hücre hastalığında nörolojik olaylar, önemli morbidite nedenidir. Çocuklarda akut büyük damar oklüzyonları meydana gelir ve tedavi edilmediğinde % 70 rekürrans oranı vardır. Bu inmeler, tekrarlayan oklüzyonların oranını azalttığı gösterilen uzun dönemli exchange transfüzyonları için bir endikasyon oluşturur. Çok iyi anlaşılamayan nedenlerle, bu tür büyük damar okı üzyonları, erişkinlerde nadiren görülür. Erişkinlerde, serebral damarlarda tekrarlayan ok1üzyonlara cevap olarak oluşan proliferatif damarların anevrizmal dilatasyonuna bağlı hemorajik inmeler görülebilir. Kemik iliğini geçici olarak baskılayan herhangi bir toksik ya da enfeksiyöz atak, ap/astik bir krize yol açabilir. Orak hücreli hastalıkta eritrositlerin kısalan ömrü, hastaları, sürekli kemik iliği aktivitesine bağımlı kıldığından, retikülosit yapımındaki düşüşler, hemoglobin ve hematokritte ciddi düşüklüklere neden olabilir. En dramatik olanı, doğrudan eritroid öncüllerinin etkilendiği parvovirüs B 19’un neden olduğu enfeksiyonlardır. Burada yapılabilecek tek şey, yalnızca destekleyici tedavidir. Ancak, bazı hastalarda lökoeritroblastik tablo ile birlikte kemik iliği nekrozu görülebilir. Bu tablo, daha sonra akciğerlerde kemik iliği embolizasyonu şeklinde komplikasyona neden olabilir. Bazı damarlar, orak hücreli hastalığın komplikasyonlarına özellikle yatkın olabilir. Renal medulla, vazooklüzyona bağlı hasara oldukça hassastır çünkü yüksek tonisite ve düşük oksijen, HbS konsantrasyonunu belirgin olarak artırır. Orak hücre hastalığı olanların hepsinde idrarı konsantre etme yeteneğinde bozukluk geliştiğinden, bu kişilerin idrarları, erişkinliğe kadar izostenürik hale gelir. Papiller nekroza sekonder akut hematüri epizodlan sık görülür. Dalak da, tekrarlayan oraklaşmanın olduğu bir organdır. Tekrarlayan mikrovasküler infarktüsler nedeniyle erişkin çağa kadar tüm hastalar, fonksiyonelolarak asplenik hale gelirler. Bu yardımcı faktörler, orak hücre hastalığı olan hastaların ankapsüle organizmalara bağlı enfeksiyonlara karşı duyarlılığını artınr. Akut enfeksiyonlar, orak hücre hastalığı olanlarda ciddi bir ölüm nedenidir. Bilinmeyen nedenlerle orak hücre hastalığı olanlar, osteomiyelite eğilimlidir, bu hastalarda osteomiyelitin sorumlu ajanı, sıklıkla Salmonella’dır.
Orak hücreli anemi, bir çocukluk hastalığı olarak bilinirdi. Giderek daha çok hastanın erişkinliğe kadar yaşamasıyla birlikte, vazooklüzyonun tekrarlayan epizodlarının yaklaşık her organda end organ hasarına yol açabileceği açıklık kazanmıştır. Orak hücreli anemisi olan erişkin hastalarda başlıca ölüm nedeni, renal ve pulmoner yetersizlikir. Diğer uzun süreli komplikasyonlar arasında, kronik deri ülserleri, retinopati ve karaciğer fonksiyon bozukluğudur. Ayrıca, pigment taşları nedeniyle pek çok hastada kolesistektonu gerekli olur.Orak hücreli anemi tedavisi büyük oranda destekleyicidir. Ağrı krizlerinin tedavisi, sıvı ve oksijen desteği ve analjeziklerdir. Enfeksiyonu olan hastalara antibiyotik verilmelidir. Semptomatik anemisi olanlara transfüzyon yapılmalıdır. Göğüs sendromu, inme, kemik iliği nekrozu ve priapizmde exchange transfüzyonu endikasyonu vardır. Exchange transfüzyon için tartışmalı endikasyonları arasında, diğer destekleyici önlemlere yavaş cevap alma ve inatçı ağrılardır. Bu transfüzyonda, %30 ve %40 arasında bir HbS düzeyi hedeflenmelidir. Daha önce de belirtildiği gibi, büyük bir damarda oluşan tromboza bağlı inme olduğunda, sürekli exchange transfüzyonu yapılmalıdır. Son çalışmalar, orak hücreli anemide Hbf konsantrasyonunu artıran bir ajan olan hidroksiüre ile tedavinin, vazookluzif krizleri insidansini azalttığını göstermektedir. Tekrarlayan krizleri olan hastalarda hidroksiürenin etkinliği, randomuze bir çalışmada gösterilmiştir, takip çalışmalarında da, hidroksiüre ile tedavi edilen hastalarda yaşam süresi avantajı gösterilmiştir. Buradaki etki, polimerizasyon olmayan BS ve bir y zincirini içeren hemoglobin tetramerleri oluşumuna atfedilmektedir (a2 Sy). Son yapılan çalışmalarda, bu tedaviye cevabın, lökosit sayısındaki azalma ve endotele yapışma özelliklerindeki değişikliklerle de bağlantılı olduğu ileri sürülmüştür.
Hemoglobin C. Hemoglobin C (HbC), a-globin zincirinin 6. pozisyonunda glutamik asidin lizin ile yer değiştirmesiyle meydana gelir. Homozigot HbC, çok hafif orak hücre semptomuna neden olur ve genellikle klinik olarak sessizdir. Hemoglobin S-C (HbSC), HbS ve HBC için bileşik heterozigottur. HbSC’li hastalar, homozigot HbS’li (HbSS) hastalardakine oranla daha hafif klinik belirti vermelerine rağmen, daha semptomatiktirler. Hastaların hematokrit düzeyi daha yüksektir ve yüksek visközite nedeniyle retinopati derecesini artınr. HbSS’li hastalardakinin aksine bu hastalarda, dalak enfarktüsleri olmaz ve splenomegali gelişir. Bu nedenle, hemoglobin ve hematokritte (dalak sekestrasyon krizleri) ciddi azalmayla birlikte akut splenomegali epizodları görülür. Her ne kadar HbSS’li çocuklarda da bu tür krizler görülebilse de, HbSS’li erişkin hastalarda bu komplikasyonu, fonksiyonel aspleni önler.